

Medical implications of technical accuracy in genome sequencing
- PMID: 26932475
- PMCID: PMC4774017
- DOI: 10.1186/s13073-016-0269-0
Medical implications of technical accuracy in genome sequencing
Abstract
Background: As whole exome sequencing (WES) and whole genome sequencing (WGS) transition from research tools to clinical diagnostic tests, it is increasingly critical for sequencing methods and analysis pipelines to be technically accurate. The Genome in a Bottle Consortium has recently published a set of benchmark SNV, indel, and homozygous reference genotypes for the pilot whole genome NIST Reference Material based on the NA12878 genome.
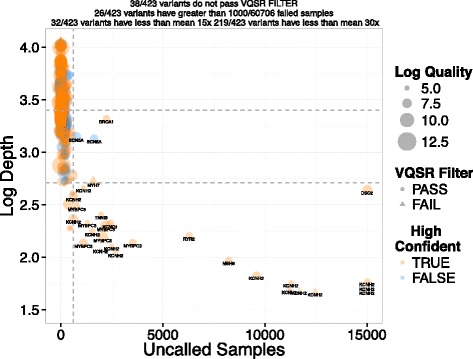
Methods: We examine the relationship between human genome complexity and genes/variants reported to be associated with human disease. Specifically, we map regions of medical relevance to benchmark regions of high or low confidence. We use benchmark data to assess the sensitivity and positive predictive value of two representative sequencing pipelines for specific classes of variation.
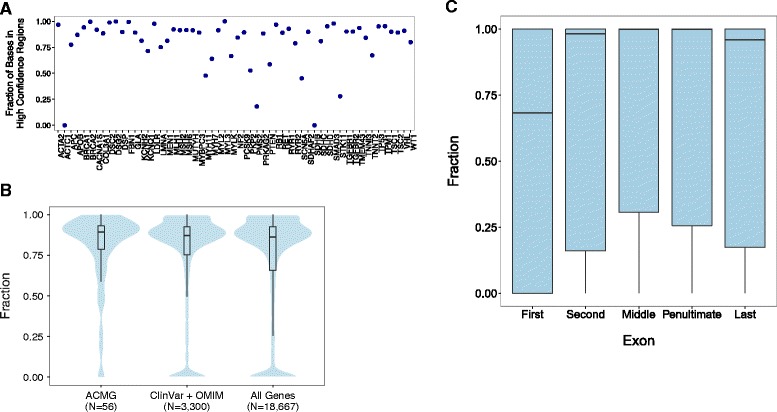
Results: We observe that the accuracy of a variant call depends on the genomic region, variant type, and read depth, and varies by analytical pipeline. We find that most false negative WGS calls result from filtering while most false negative WES variants relate to poor coverage. We find that only 74.6% of the exonic bases in ClinVar and OMIM genes and 82.1% of the exonic bases in ACMG-reportable genes are found in high-confidence regions. Only 990 genes in the genome are found entirely within high-confidence regions while 593 of 3,300 ClinVar/OMIM genes have less than 50% of their total exonic base pairs in high-confidence regions. We find greater than 77 % of the pathogenic or likely pathogenic SNVs currently in ClinVar fall within high-confidence regions. We identify sites that are prone to sequencing errors, including thousands present in publicly available variant databases. Finally, we examine the clinical impact of mandatory reporting of secondary findings, highlighting a false positive variant found in BRCA2.
Conclusions: Together, these data illustrate the importance of appropriate use and continued improvement of technical benchmarks to ensure accurate and judicious interpretation of next-generation DNA sequencing results in the clinical setting.
Figures
Comment in
-
Genetic testing: Clinical sequencing right on target.Nat Rev Genet. 2016 May;17(5):253. doi: 10.1038/nrg.2016.34. Epub 2016 Mar 21. Nat Rev Genet. 2016. PMID: 26996078 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
