

ViromeScan: a new tool for metagenomic viral community profiling
- PMID: 26932765
- PMCID: PMC4774116
- DOI: 10.1186/s12864-016-2446-3
ViromeScan: a new tool for metagenomic viral community profiling
Abstract
Background: Bioinformatics tools available for metagenomic sequencing analysis are principally devoted to the identification of microorganisms populating an ecological niche, but they usually do not consider viruses. Only some software have been designed to profile the viral sequences, however they are not efficient in the characterization of viruses in the context of complex communities, like the intestinal microbiota, containing bacteria, archeabacteria, eukaryotic microorganisms and viruses. In any case, a comprehensive description of the host-microbiota interactions can not ignore the profile of eukaryotic viruses within the virome, as viruses are definitely critical for the regulation of the host immunophenotype.
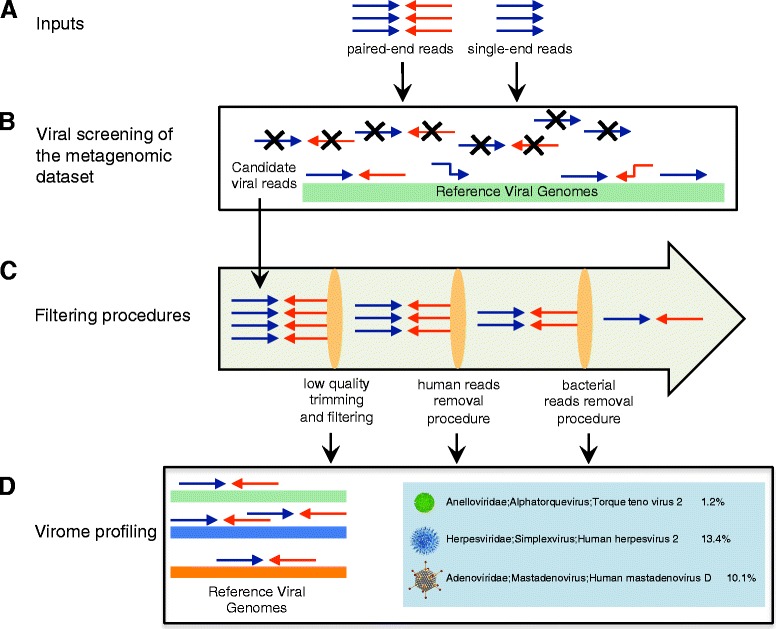
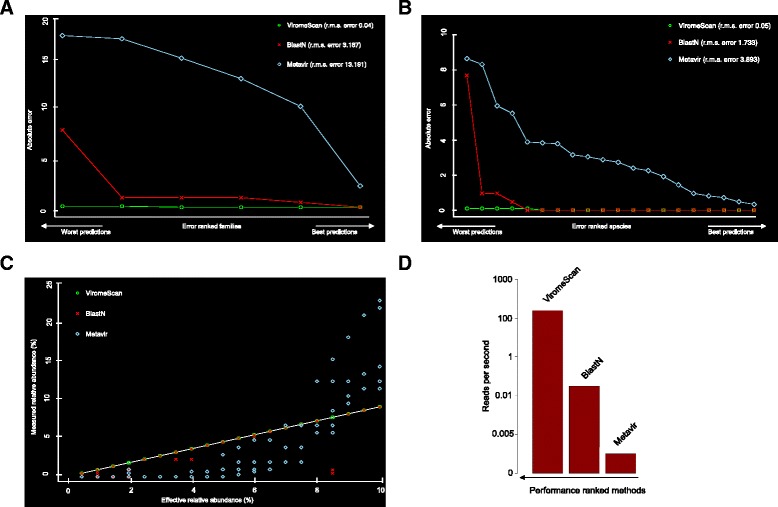
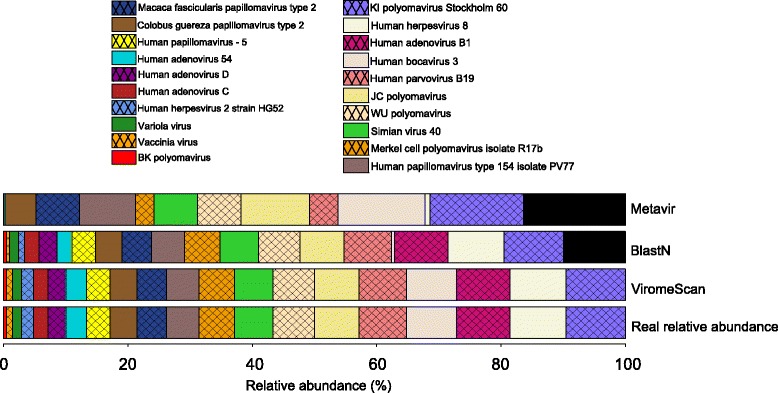
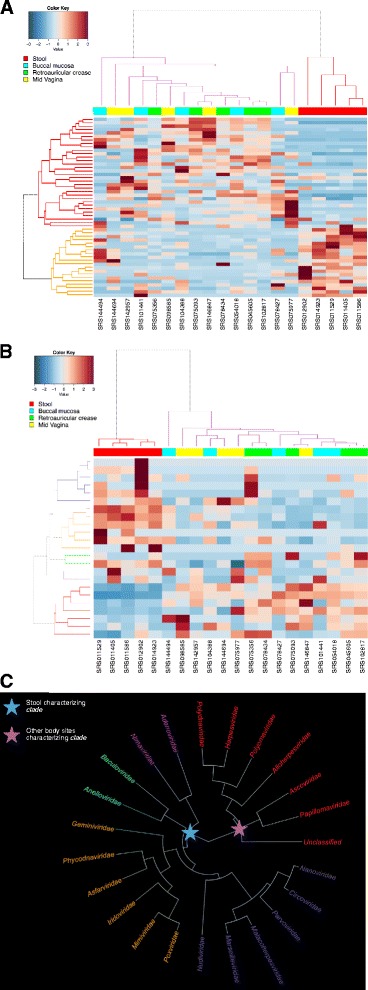
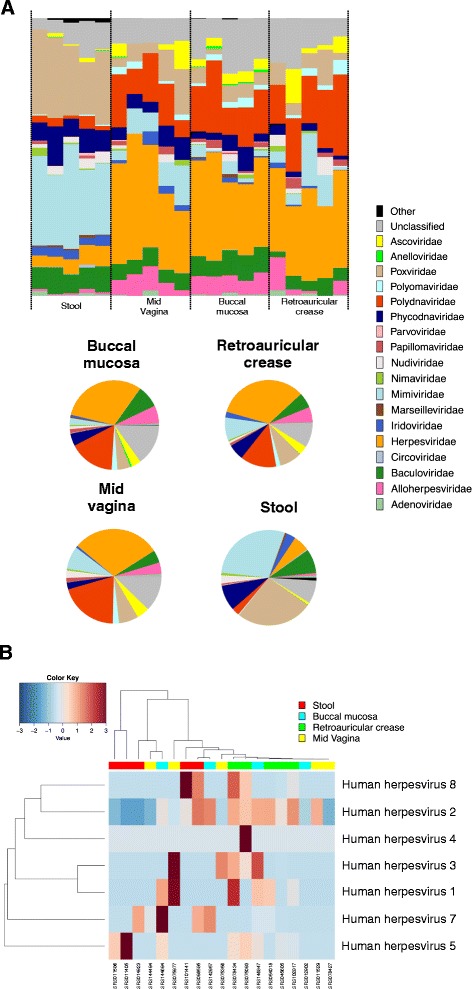
Results: ViromeScan is an innovative metagenomic analysis tool that characterizes the taxonomy of the virome directly from raw data of next-generation sequencing. The tool uses hierarchical databases for eukaryotic viruses to unambiguously assign reads to viral species more accurately and >1000 fold faster than other existing approaches. We validated ViromeScan on synthetic microbial communities and applied it on metagenomic samples of the Human Microbiome Project, providing a sensitive eukaryotic virome profiling of different human body sites.
Conclusions: ViromeScan allows the user to explore and taxonomically characterize the virome from metagenomic reads, efficiently denoising samples from reads of other microorganisms. This implies that users can fully characterize the microbiome, including bacteria and viruses, by shotgun metagenomic sequencing followed by different bioinformatic pipelines.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
