

Pre-fusion structure of a human coronavirus spike protein
- PMID: 26935699
- PMCID: PMC4860016
- DOI: 10.1038/nature17200
Pre-fusion structure of a human coronavirus spike protein
Abstract
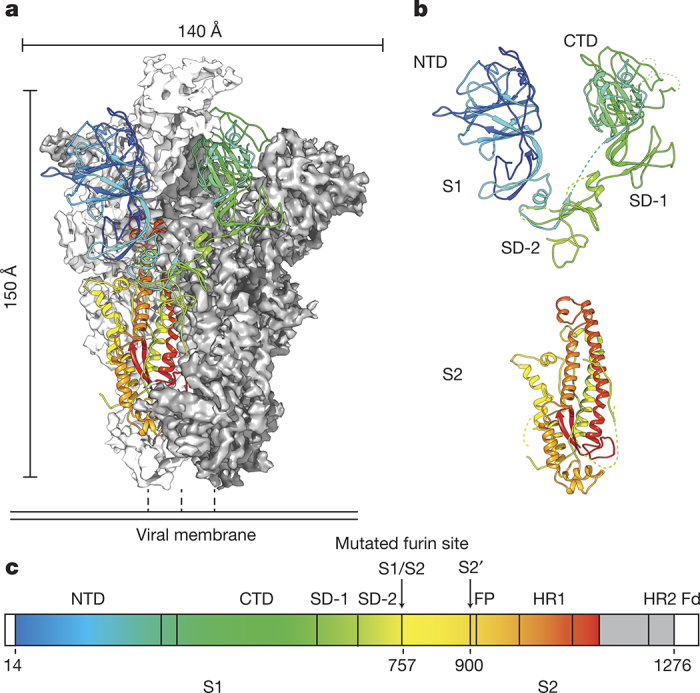
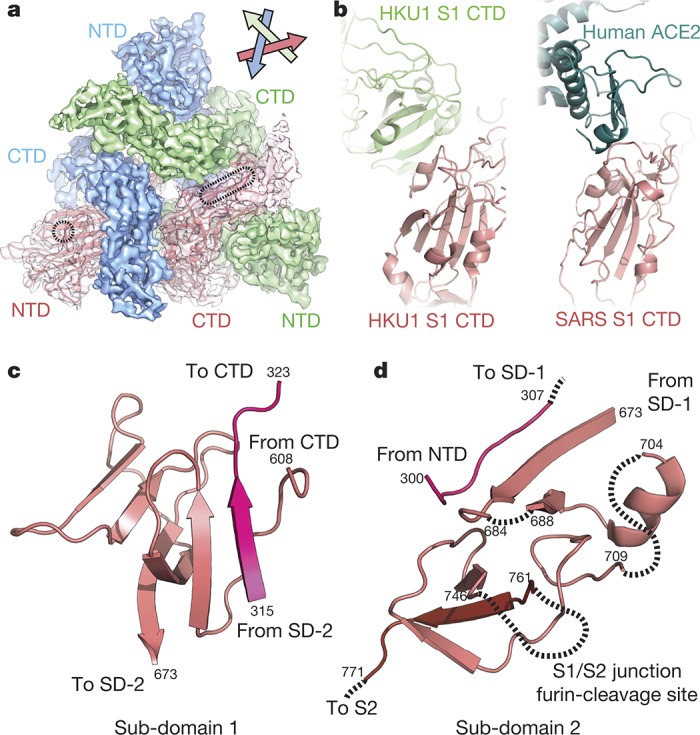
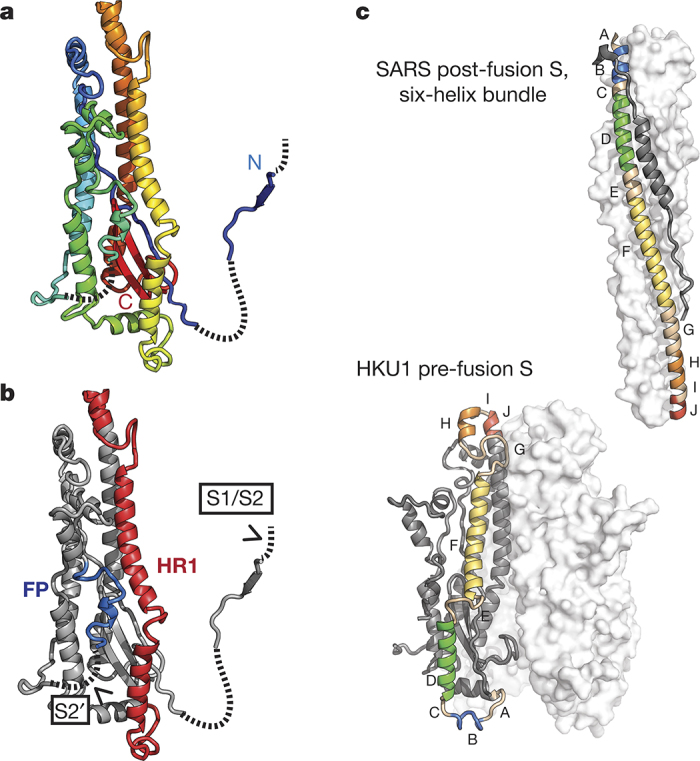
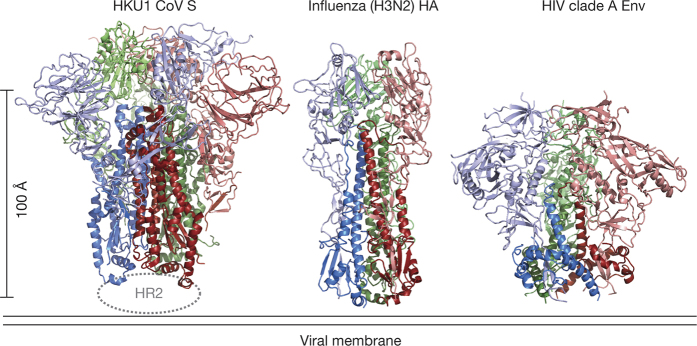
HKU1 is a human betacoronavirus that causes mild yet prevalent respiratory disease, and is related to the zoonotic SARS and MERS betacoronaviruses, which have high fatality rates and pandemic potential. Cell tropism and host range is determined in part by the coronavirus spike (S) protein, which binds cellular receptors and mediates membrane fusion. As the largest known class I fusion protein, its size and extensive glycosylation have hindered structural studies of the full ectodomain, thus preventing a molecular understanding of its function and limiting development of effective interventions. Here we present the 4.0 Å resolution structure of the trimeric HKU1 S protein determined using single-particle cryo-electron microscopy. In the pre-fusion conformation, the receptor-binding subunits, S1, rest above the fusion-mediating subunits, S2, preventing their conformational rearrangement. Surprisingly, the S1 C-terminal domains are interdigitated and form extensive quaternary interactions that occlude surfaces known in other coronaviruses to bind protein receptors. These features, along with the location of the two protease sites known to be important for coronavirus entry, provide a structural basis to support a model of membrane fusion mediated by progressive S protein destabilization through receptor binding and proteolytic cleavage. These studies should also serve as a foundation for the structure-based design of betacoronavirus vaccine immunogens.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
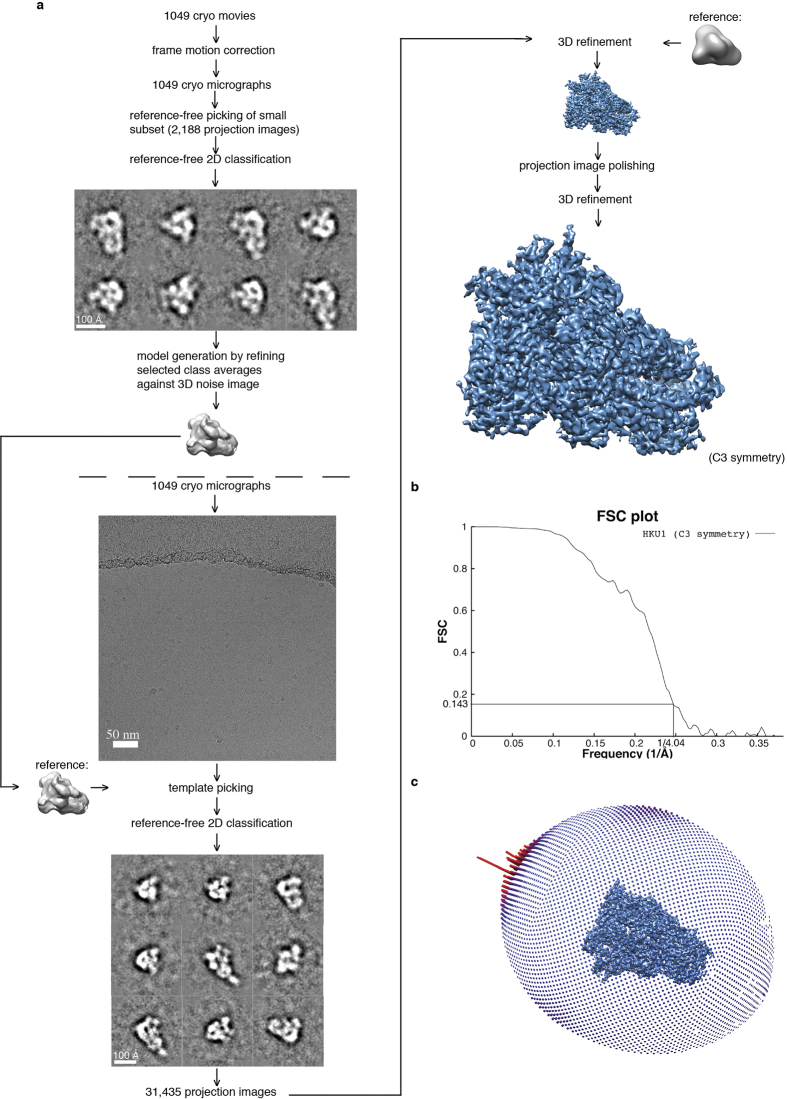
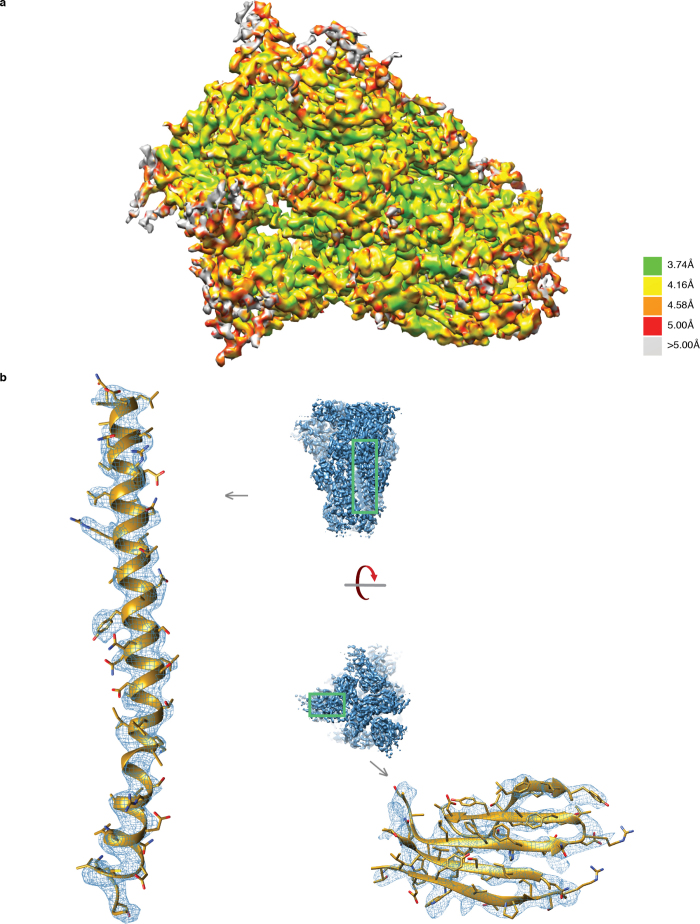
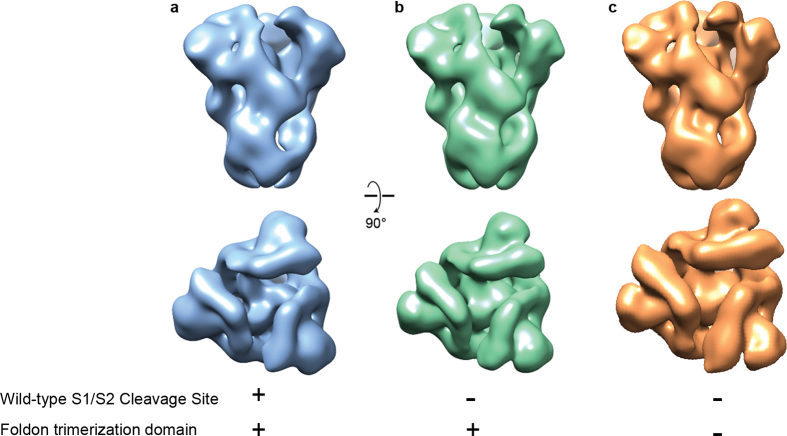
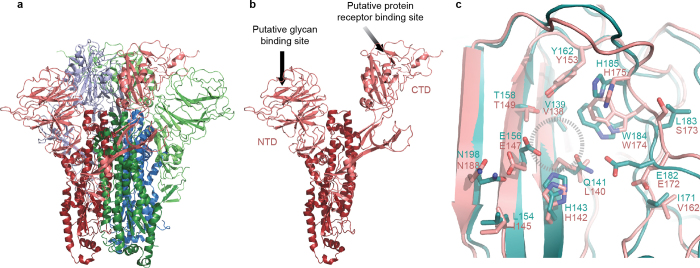
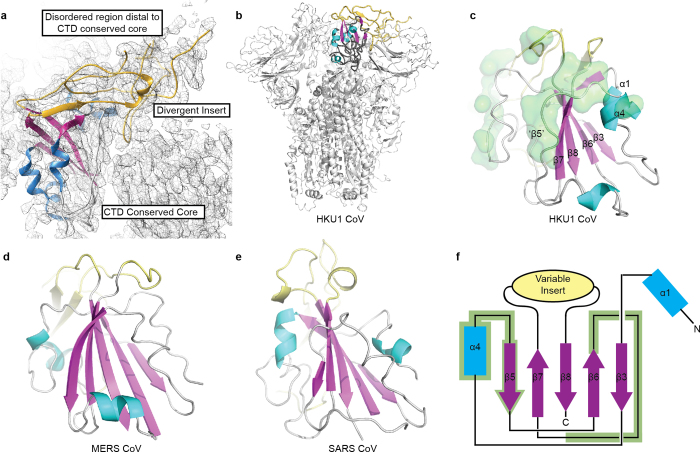

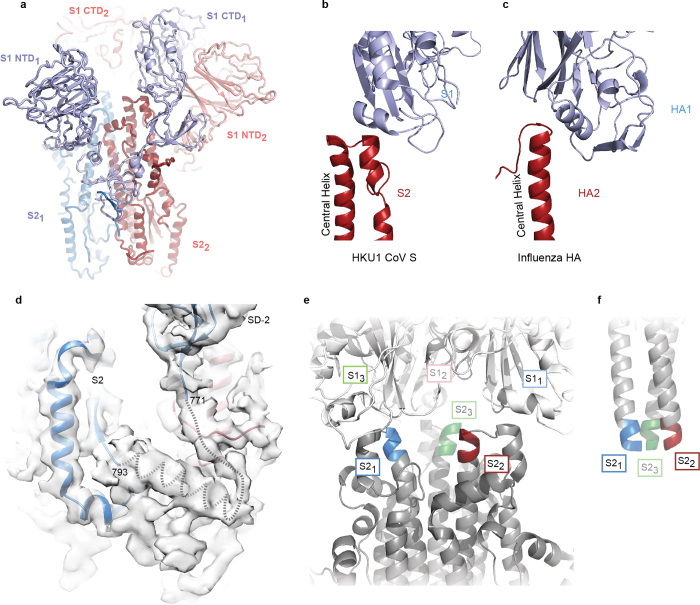
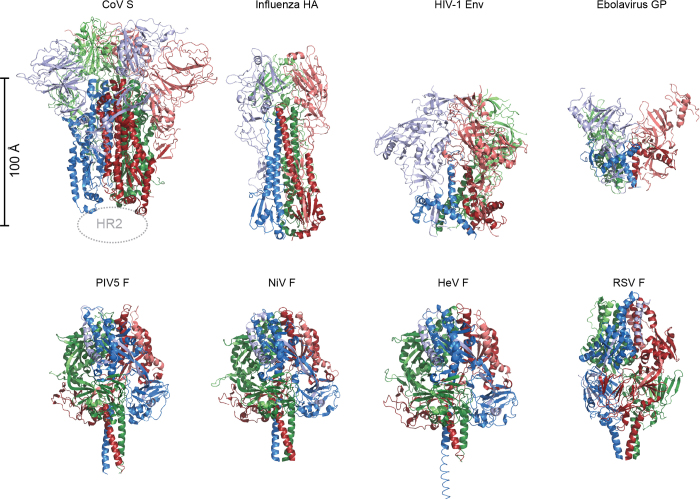
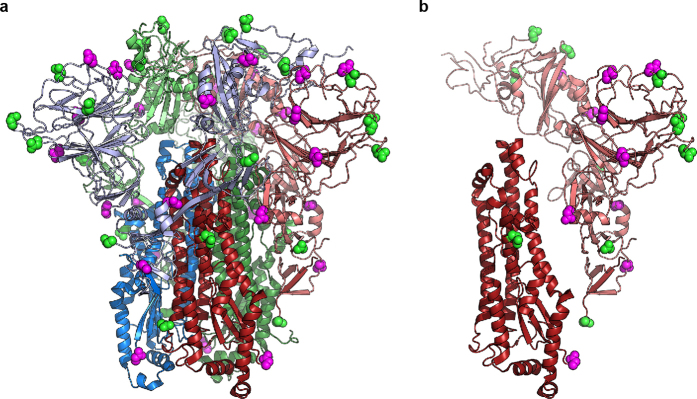
References
-
- Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
