

Minireview: Genetic basis of heterogeneity and severity in sickle cell disease
- PMID: 26936084
- PMCID: PMC4950383
- DOI: 10.1177/1535370216636726
Minireview: Genetic basis of heterogeneity and severity in sickle cell disease
Abstract
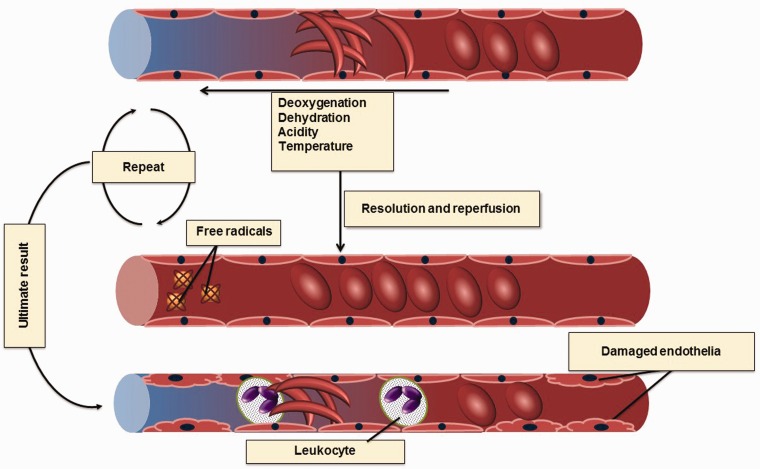
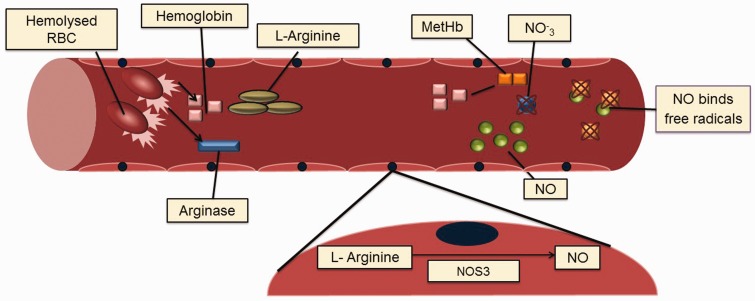
Sickle cell disease, a common single gene disorder, has a complex pathophysiology that at its root is initiated by the polymerization of deoxy sickle hemoglobin. Sickle vasoocclusion and hemolytic anemia drive the development of disease complications. In this review, we focus on the genetic modifiers of disease heterogeneity. The phenotypic heterogeneity of disease is only partially explained by genetic variability of fetal hemoglobin gene expression and co-inheritance of α thalassemia. Given the complexity of pathophysiology, many different definitions of severity are possible complicating a full understanding of its genetic foundation. The pathophysiological complexity and the interlocking nature of the biological processes underpinning disease severity are becoming better understood. Nevertheless, useful genetic signatures of severity, regardless of how this is defined, are insufficiently developed to be used for treatment decisions and for counseling.
Keywords: Severity in sickle cell disease; genome-wide association study; genotype–phenotype correlation; hemolysis; single nucleotide polymorphisms; subphenotypes of sickle cell disease.
© 2016 by the Society for Experimental Biology and Medicine.
Figures
References
-
- Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. 2009; 94: 602–603.
-
- Ranney HM. Historical milestones. In: Embury SH, Hebbel RP, Mohandas N, Steinberg MH. (eds). Sickle cell disease: basic principles and clinical practice, New York, NY: Lippincott-Raven, 1994, pp. 1–5.
-
- Nouraie M, Lee JS, Zhang Y, Kanias T, Zhao X, Xiong Z, Oriss TB, Zeng Q, Kato GJ, Gibbs JS, Hildesheim ME. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013; 98: 464–72. - PMC - PubMed
-
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010; 376: 2018–31. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical