

Cardiac Stim1 Silencing Impairs Adaptive Hypertrophy and Promotes Heart Failure Through Inactivation of mTORC2/Akt Signaling
- PMID: 26936863
- PMCID: PMC4829441
- DOI: 10.1161/CIRCULATIONAHA.115.020678
Cardiac Stim1 Silencing Impairs Adaptive Hypertrophy and Promotes Heart Failure Through Inactivation of mTORC2/Akt Signaling
Abstract
Background: Stromal interaction molecule 1 (STIM1) is a dynamic calcium signal transducer implicated in hypertrophic growth of cardiomyocytes. STIM1 is thought to act as an initiator of cardiac hypertrophic response at the level of the sarcolemma, but the pathways underpinning this effect have not been examined.
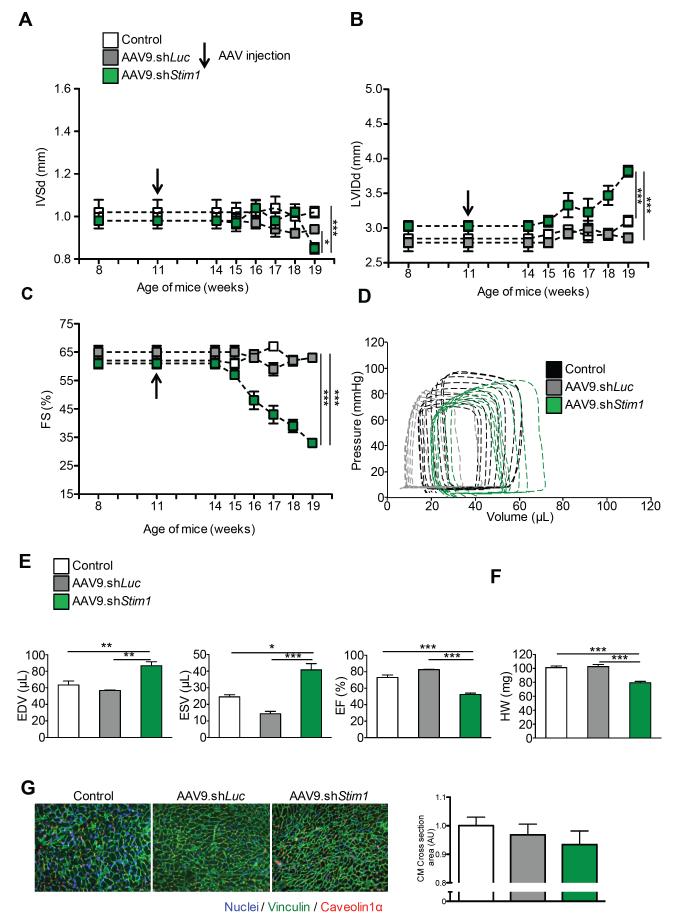
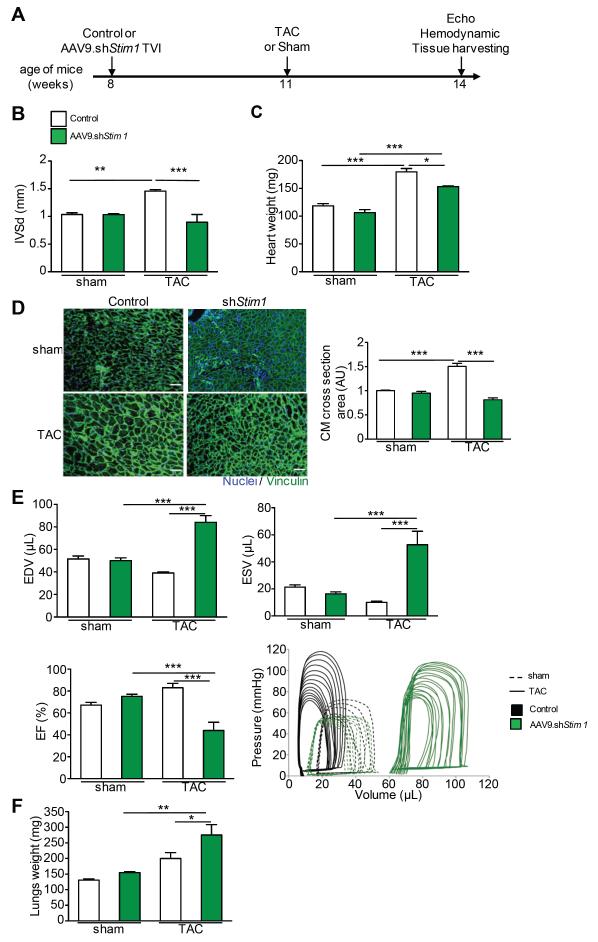
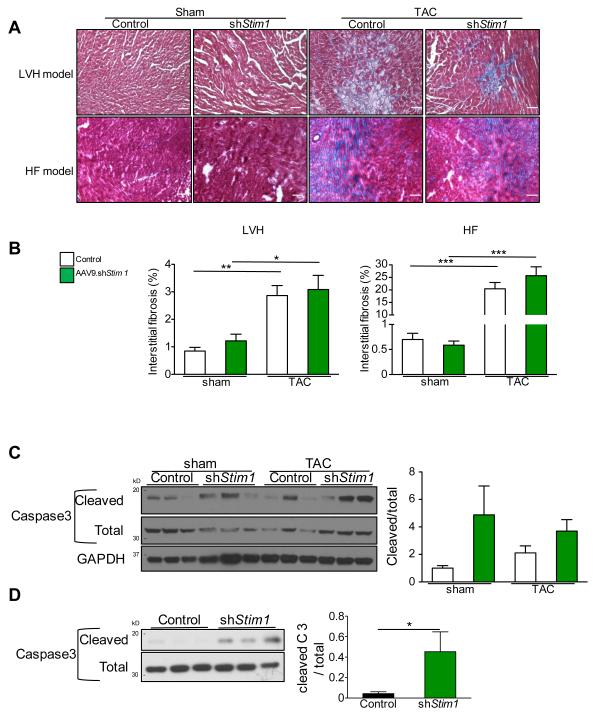
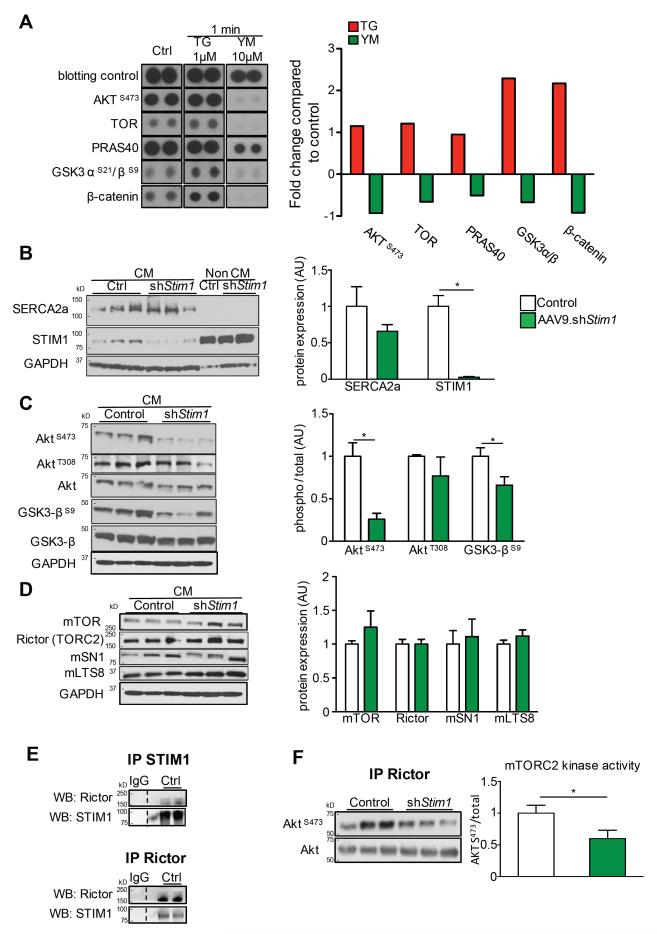
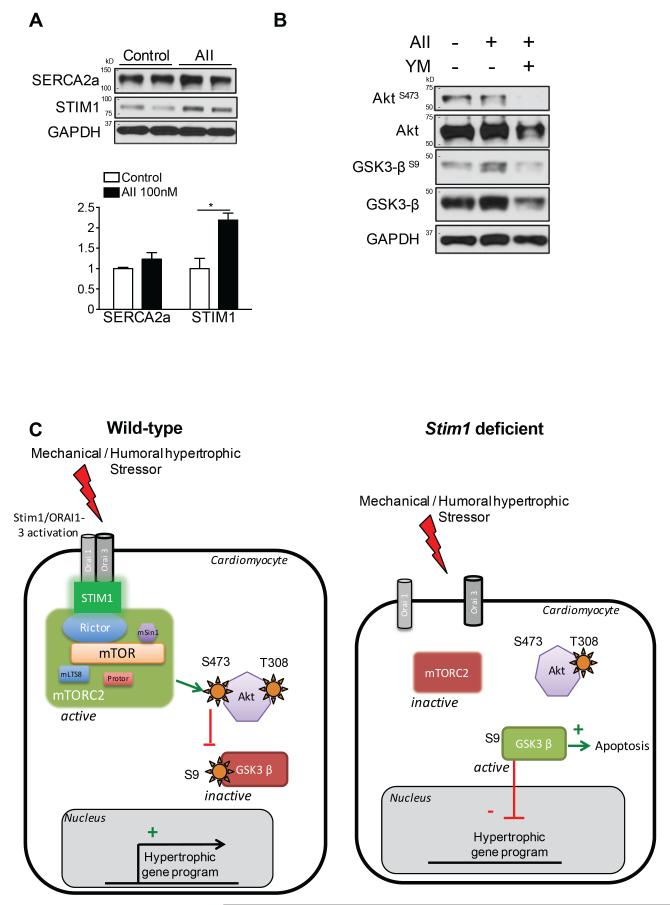
Methods and results: To determine the mechanistic role of STIM1 in cardiac hypertrophy and during the transition to heart failure, we manipulated STIM1 expression in mice cardiomyocytes by using in vivo gene delivery of specific short hairpin RNAs. In 3 different models, we found that Stim1 silencing prevents the development of pressure overload-induced hypertrophy but also reverses preestablished cardiac hypertrophy. Reduction in STIM1 expression promoted a rapid transition to heart failure. We further showed that Stim1 silencing resulted in enhanced activity of the antihypertrophic and proapoptotic GSK-3β molecule. Pharmacological inhibition of glycogen synthase kinase-3 was sufficient to reverse the cardiac phenotype observed after Stim1 silencing. At the level of ventricular myocytes, Stim1 silencing or inhibition abrogated the capacity for phosphorylation of Akt(S473), a hydrophobic motif of Akt that is directly phosphorylated by mTOR complex 2. We found that Stim1 silencing directly impaired mTOR complex 2 kinase activity, which was supported by a direct interaction between STIM1 and Rictor, a specific component of mTOR complex 2.
Conclusions: These data support a model whereby STIM1 is critical to deactivate a key negative regulator of cardiac hypertrophy. In cardiomyocytes, STIM1 acts by tuning Akt kinase activity through activation of mTOR complex 2, which further results in repression of GSK-3β activity.
Keywords: Stim1 protein, mouse; TOR complex 2; calcium; genetic therapy; heart failure.
© 2016 American Heart Association, Inc.
Figures


References
-
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. - PubMed
-
- Hulot JS, Fauconnier J, Ramanujam D, Chaanine A, Aubart F, Sassi Y, Merkle S, Cazorla O, Ouille A, Dupuis M, Hadri L, Jeong D, Muhlstedt S, Schmitt J, Braun A, Benard L, Saliba Y, Laggerbauer B, Nieswandt B, Lacampagne A, Hajjar RJ, Lompre AM, Engelhardt S. Critical role for stromal interaction molecule 1 in cardiac hypertrophy. Circulation. 2011;124:796–805. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HHSN268201000045C/HL/NHLBI NIH HHS/United States
- HL093183/HL/NHLBI NIH HHS/United States
- HHSN268201000045C/PHS HHS/United States
- R01 HL119046/HL/NHLBI NIH HHS/United States
- R01 HL128099/HL/NHLBI NIH HHS/United States
- R00HL116645/HL/NHLBI NIH HHS/United States
- P20HL100396/HL/NHLBI NIH HHS/United States
- R00 HL116645/HL/NHLBI NIH HHS/United States
- R01 HL113497/HL/NHLBI NIH HHS/United States
- R01 HL128072/HL/NHLBI NIH HHS/United States
- P20 HL100396/HL/NHLBI NIH HHS/United States
- HL083156/HL/NHLBI NIH HHS/United States
- R01 HL083156/HL/NHLBI NIH HHS/United States
- HL119046/HL/NHLBI NIH HHS/United States
- R01 HL093183/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
