

UVnovo: A de Novo Sequencing Algorithm Using Single Series of Fragment Ions via Chromophore Tagging and 351 nm Ultraviolet Photodissociation Mass Spectrometry
- PMID: 26938041
- PMCID: PMC4850734
- DOI: 10.1021/acs.analchem.6b00261
UVnovo: A de Novo Sequencing Algorithm Using Single Series of Fragment Ions via Chromophore Tagging and 351 nm Ultraviolet Photodissociation Mass Spectrometry
Abstract
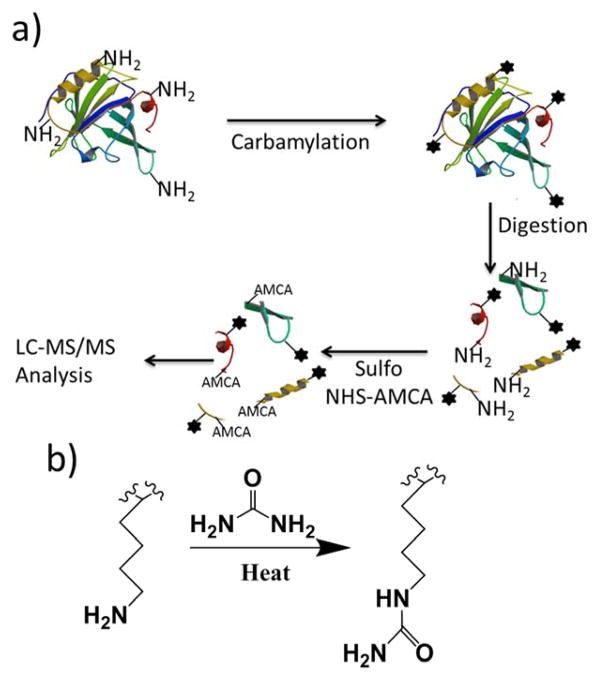
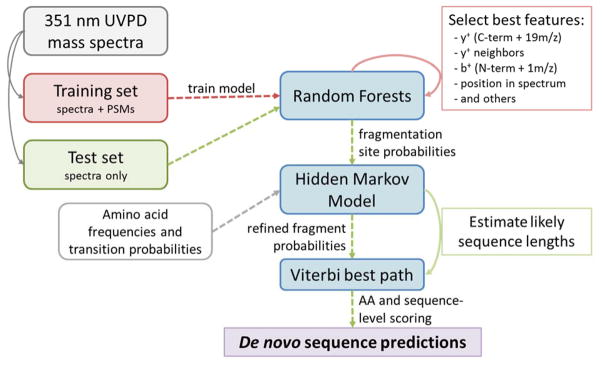
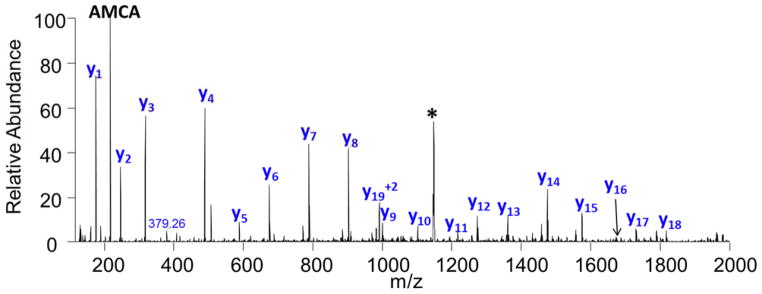
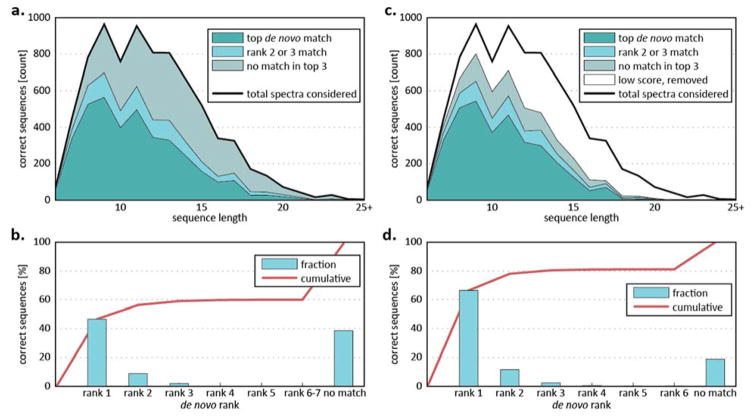
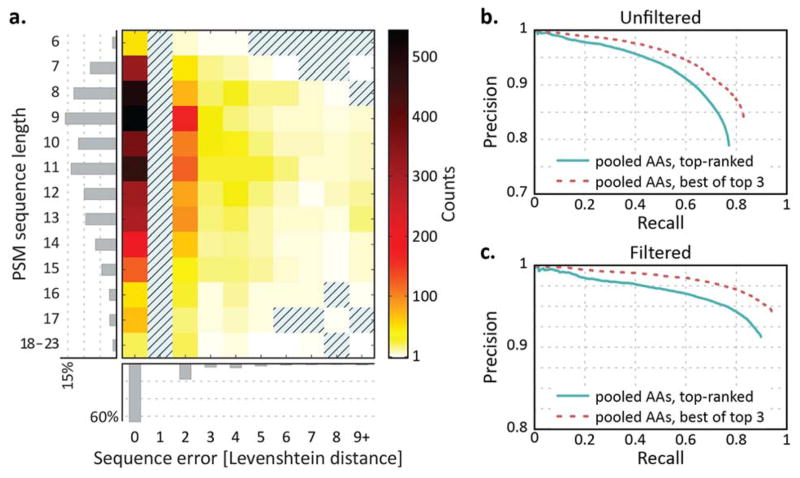
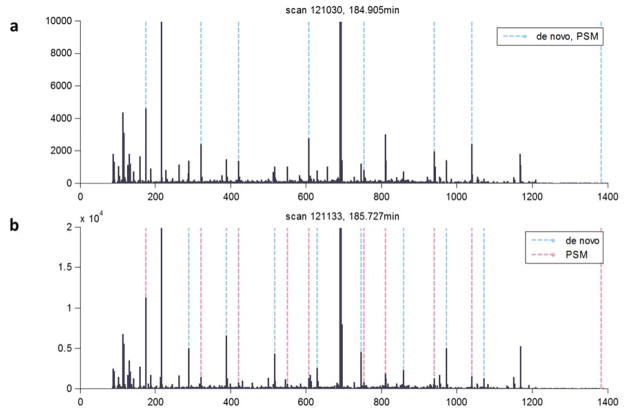
De novo peptide sequencing by mass spectrometry represents an important strategy for characterizing novel peptides and proteins, in which a peptide's amino acid sequence is inferred directly from the precursor peptide mass and tandem mass spectrum (MS/MS or MS(3)) fragment ions, without comparison to a reference proteome. This method is ideal for organisms or samples lacking a complete or well-annotated reference sequence set. One of the major barriers to de novo spectral interpretation arises from confusion of N- and C-terminal ion series due to the symmetry between b and y ion pairs created by collisional activation methods (or c, z ions for electron-based activation methods). This is known as the "antisymmetric path problem" and leads to inverted amino acid subsequences within a de novo reconstruction. Here, we combine several key strategies for de novo peptide sequencing into a single high-throughput pipeline: high-efficiency carbamylation blocks lysine side chains, and subsequent tryptic digestion and N-terminal peptide derivatization with the ultraviolet chromophore AMCA yield peptides susceptible to 351 nm ultraviolet photodissociation (UVPD). UVPD-MS/MS of the AMCA-modified peptides then predominantly produces y ions in the MS/MS spectra, specifically addressing the antisymmetric path problem. Finally, the program UVnovo applies a random forest algorithm to automatically learn from and then interpret UVPD mass spectra, passing results to a hidden Markov model for de novo sequence prediction and scoring. We show this combined strategy provides high-performance de novo peptide sequencing, enabling the de novo sequencing of thousands of peptides from an Escherichia coli lysate at high confidence.
Figures
References
-
- Seidler J, Zinn N, Boehm ME, Lehmann WD. PROTEOMICS. 2010;10(4):634–649. - PubMed
-
- Mitchell Wells J, McLuckey SA. In: Methods in Enzymology. Burlingame AL, editor. Vol. 402. Academic Press; 2005. pp. 148–185. - PubMed
-
- Laskin J, Futrell JH. Mass Spectrom Rev. 2003;22(3):158–181. - PubMed
-
- Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Nat Meth. 2007;4(9):709–712. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
