

Intra-epidemic evolutionary dynamics of a Dengue virus type 1 population reveal mutant spectra that correlate with disease transmission
- PMID: 26940650
- PMCID: PMC4778070
- DOI: 10.1038/srep22592
Intra-epidemic evolutionary dynamics of a Dengue virus type 1 population reveal mutant spectra that correlate with disease transmission
Abstract
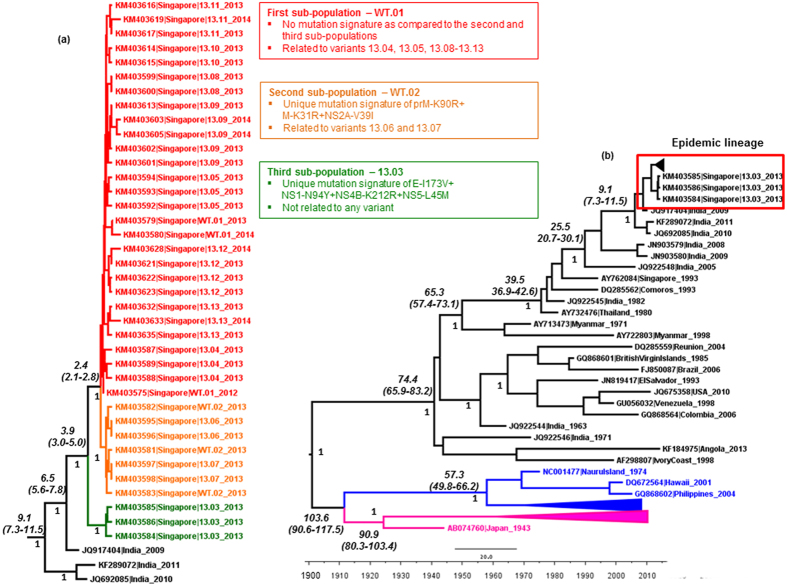
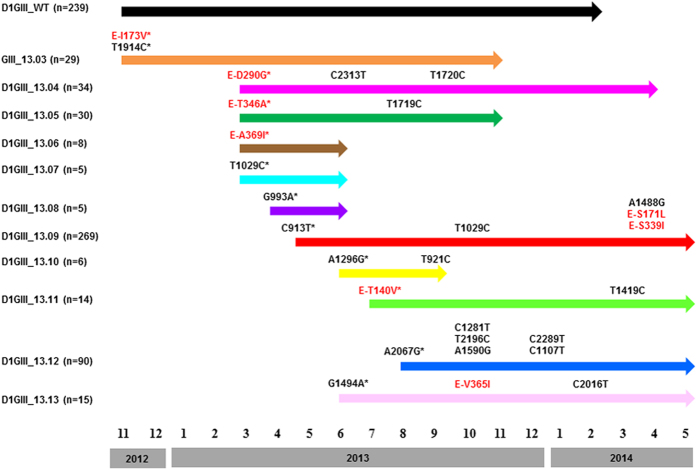
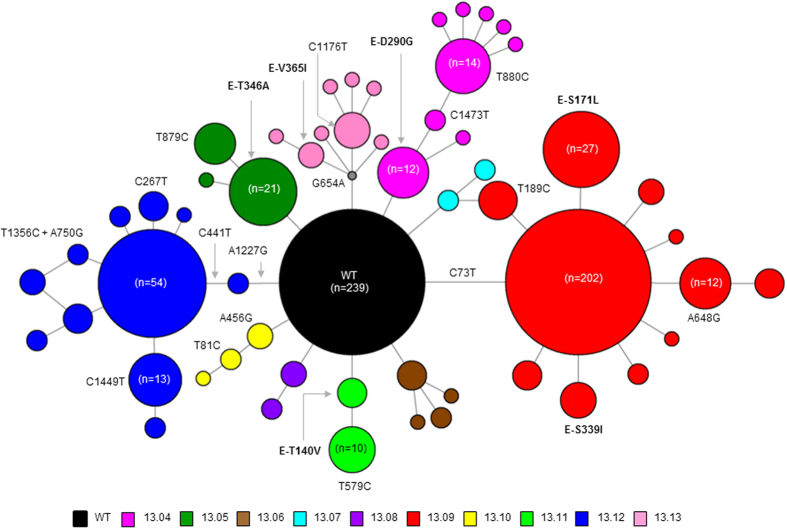
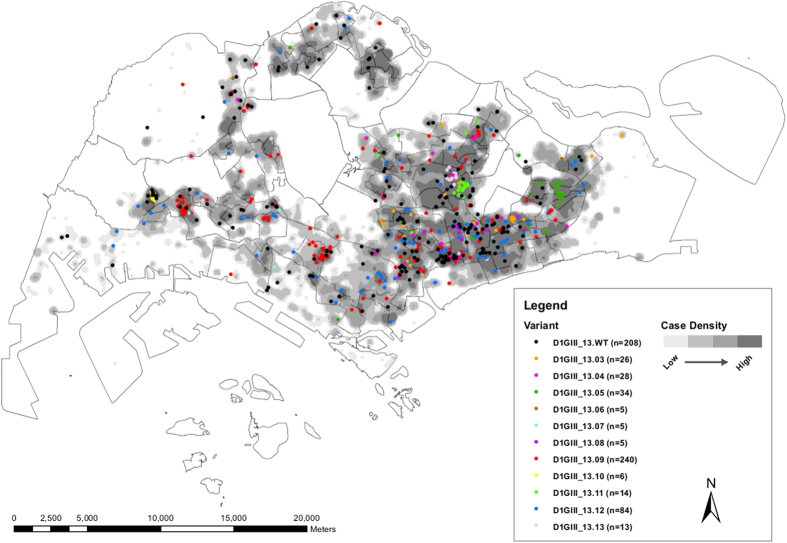
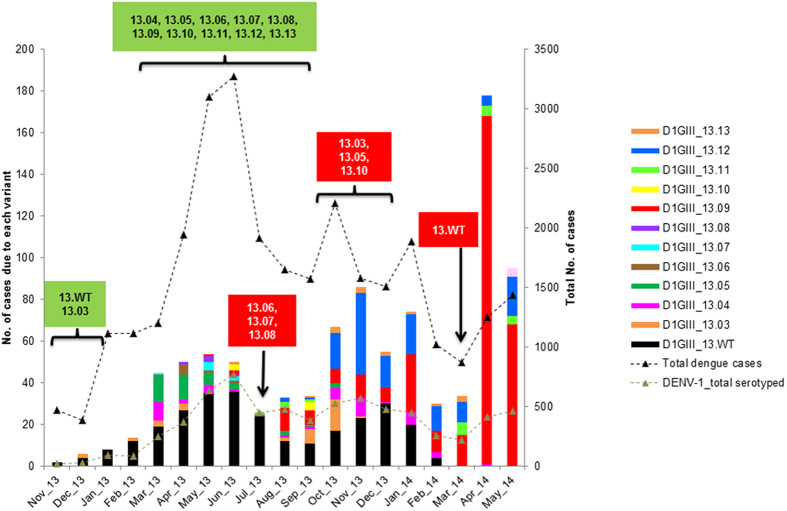
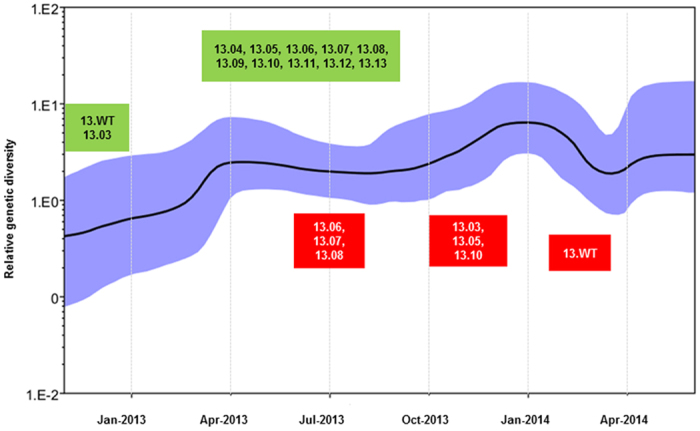
Dengue virus (DENV) is currently the most prevalent mosquito-borne viral pathogen. DENVs naturally exist as highly heterogeneous populations. Even though the descriptions on DENV diversity are plentiful, only a few studies have narrated the dynamics of intra-epidemic virus diversity at a fine scale. Such accounts are important to decipher the reciprocal relationship between viral evolutionary dynamics and disease transmission that shape dengue epidemiology. In the current study, we present a micro-scale genetic analysis of a monophyletic lineage of DENV-1 genotype III (epidemic lineage) detected from November 2012 to May 2014. The lineage was involved in an unprecedented dengue epidemic in Singapore during 2013-2014. Our findings showed that the epidemic lineage was an ensemble of mutants (variants) originated from an initial mixed viral population. The composition of mutant spectrum was dynamic and positively correlated with case load. The close interaction between viral evolution and transmission intensity indicated that tracking genetic diversity through time is potentially a useful tool to infer DENV transmission dynamics and thereby, to assess the epidemic risk in a disease control perspective. Moreover, such information is salient to understand the viral basis of clinical outcome and immune response variations that is imperative to effective vaccine design.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
