

Accelerated geroncogenesis in hereditary breast-ovarian cancer syndrome
- PMID: 26943589
- PMCID: PMC4914261
- DOI: 10.18632/oncotarget.7867
Accelerated geroncogenesis in hereditary breast-ovarian cancer syndrome
Abstract
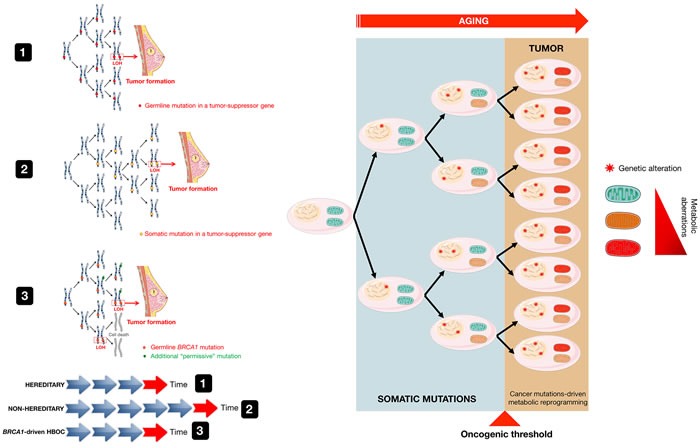
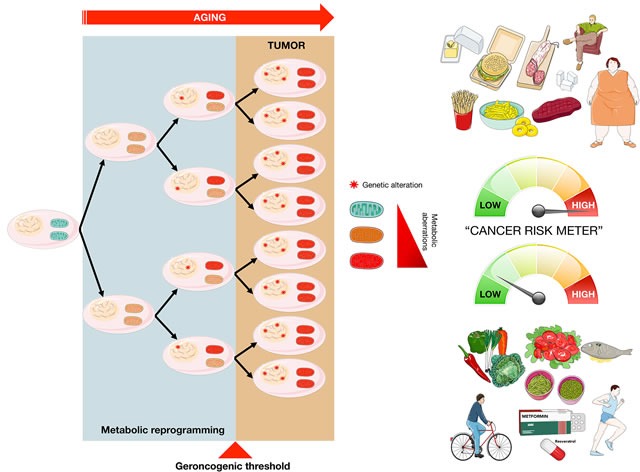
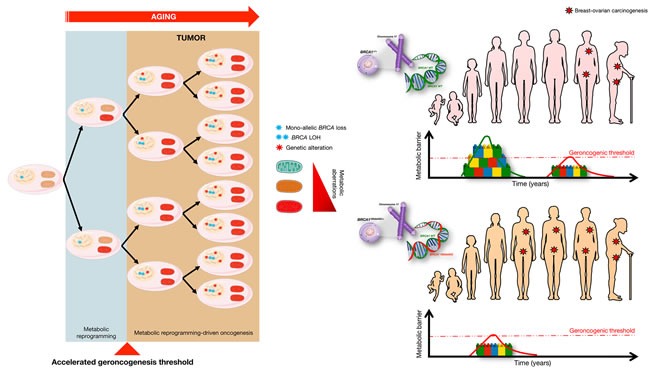
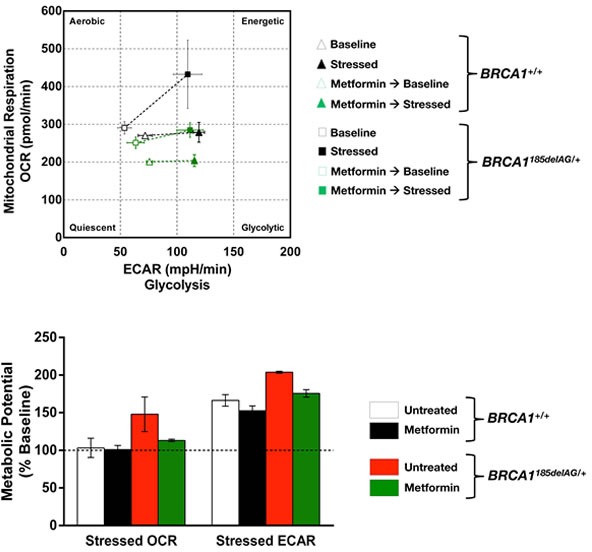
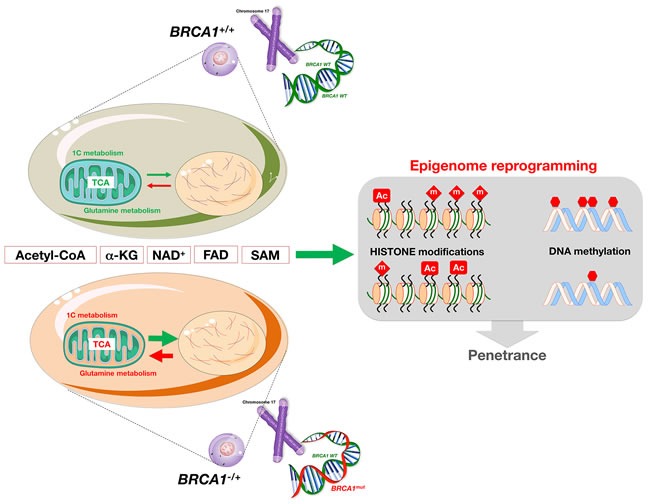
The geroncogenesis hypothesis postulates that the decline in metabolic cellular health that occurs naturally with aging drives a "field effect" predisposing normal tissues for cancer development. We propose that mutations in the cancer susceptibility genes BRCA1/2 might trigger "accelerated geroncogenesis" in breast and ovarian epithelia. By speeding up the rate at which the metabolic threshold becomes "permissive" with survival and expansion of genomically unstable pre-tumoral epithelial cells, BRCA haploinsufficiency-driven metabolic reprogramming would operate as a bona fide oncogenic event enabling malignant transformation and tumor formation in BRCA carriers. The metabolic facet of BRCA1 one-hit might involve tissue-specific alterations in acetyl-CoA, α-ketoglutarate, NAD+, FAD, or S-adenosylmethionine, critical factors for de/methylation or de/acetylation dynamics in the nuclear epigenome. This in turn might induce faulty epigenetic reprogramming at the "install phase" that directs cell-specific differentiation of breast/ovarian epithelial cells, which can ultimately determine the penetrance of BRCA defects during developmental windows of susceptibility. This model offers a framework to study whether metabolic drugs that prevent or revert metabolic reprogramming induced by BRCA haploinsufficiency might displace the "geroncogenic risk" of BRCA carriers to the age typical for those without the mutation. The identification of the key nodes that directly communicate changes in cellular metabolism to the chromatin in BRCA haploinsufficient cells may allow the epigenetic targeting of genomic instability using exclusively metabolic means. The validation of accelerated geroncogenesis as an inherited "one-hit" metabolic "field effect" might offer new strategies to therapeutically revisit the apparently irreversible genetic-hereditary fate of women with hereditary breast-ovarian cancer syndrome.
Keywords: BRCA1; Gerotarget; cancer; geroncogenesis; metabolism; metformin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Pathologic findings in breast, fallopian tube, and ovary specimens in non-BRCA hereditary breast and/or ovarian cancer syndromes: a study of 18 patients with deleterious germline mutations in RAD51C, BARD1, BRIP1, PALB2, MUTYH, or CHEK2.Hum Pathol. 2017 Dec;70:14-26. doi: 10.1016/j.humpath.2017.06.018. Epub 2017 Jul 12. Hum Pathol. 2017. PMID: 28709830
-
Hereditary cancer-associated mutations in women diagnosed with two primary cancers: an opportunity to identify hereditary cancer syndromes after the first cancer diagnosis.Oncology. 2015;88(4):226-33. doi: 10.1159/000368836. Epub 2014 Dec 11. Oncology. 2015. PMID: 25503195
-
Germline BRCA1 mutation reprograms breast epithelial cell metabolism towards mitochondrial-dependent biosynthesis: evidence for metformin-based "starvation" strategies in BRCA1 carriers.Oncotarget. 2016 Aug 16;7(33):52974-52992. doi: 10.18632/oncotarget.9732. Oncotarget. 2016. PMID: 27259235 Free PMC article.
-
Hereditary breast-ovarian cancer: clinical findings and medical management.Plast Surg Nurs. 2007 Jul-Sep;27(3):124-7. doi: 10.1097/01.PSN.0000290280.48197.e7. Plast Surg Nurs. 2007. PMID: 17901820 Review.
-
Hereditary ovarian cancer.Crit Rev Oncol Hematol. 2009 Jan;69(1):28-44. doi: 10.1016/j.critrevonc.2008.06.003. Epub 2008 Jul 24. Crit Rev Oncol Hematol. 2009. PMID: 18656380 Review.
Cited by
-
RAD51 and PALB2 in precision oncology: Clinical implications for HRD associated breast and ovarian cancers (Review).Int J Oncol. 2025 Aug;67(2):65. doi: 10.3892/ijo.2025.5771. Epub 2025 Jul 4. Int J Oncol. 2025. PMID: 40613200 Free PMC article. Review.
-
Metformin inhibits RANKL and sensitizes cancer stem cells to denosumab.Cell Cycle. 2017 Jun 3;16(11):1022-1028. doi: 10.1080/15384101.2017.1310353. Epub 2017 Apr 7. Cell Cycle. 2017. PMID: 28387573 Free PMC article.
-
Metformin targets histone acetylation in cancer-prone epithelial cells.Cell Cycle. 2016 Dec 16;15(24):3355-3361. doi: 10.1080/15384101.2016.1249547. Epub 2016 Oct 28. Cell Cycle. 2016. PMID: 27792453 Free PMC article.
-
Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism.Oncogene. 2018 Feb 15;37(7):963-970. doi: 10.1038/onc.2017.367. Epub 2017 Oct 23. Oncogene. 2018. PMID: 29059169
-
BRCA1 haploinsufficiency cell-autonomously activates RANKL expression and generates denosumab-responsive breast cancer-initiating cells.Oncotarget. 2017 May 23;8(21):35019-35032. doi: 10.18632/oncotarget.16558. Oncotarget. 2017. PMID: 28388533 Free PMC article.
References
-
- Evans DG, Barwell J, Eccles DM, Collins A, Izatt L, Jacobs C, Donaldson A, Brady AF, Cuthbert A, Harrison R, Thomas S, Howell A, FH02 Study Group. RGC teams. Miedzybrodzka Z, Murray A. The Angelina Jolie effect: how high celebrity profile can have a major impact on provision of cancer related services. Breast Cancer Res. 2014;16:442. - PMC - PubMed
-
- Kamenova K, Reshef A, Caulfield T. Angelina Jolie's faulty gene: newspaper coverage of a celebrity's preventive bilateral mastectomy in Canada, the United States, and the United Kingdom. Genet Med. 2014;16:522–8. - PubMed
-
- Lebo PB, Quehenberger F, Kamolz LP, Lumenta DB. The Angelina effect revisited: Exploring a media-related impact on public awareness. Cancer. 2015;121:3959–3964. - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
