

The Most Prevalent Freeman-Sheldon Syndrome Mutations in the Embryonic Myosin Motor Share Functional Defects
- PMID: 26945064
- PMCID: PMC4858979
- DOI: 10.1074/jbc.M115.707489
The Most Prevalent Freeman-Sheldon Syndrome Mutations in the Embryonic Myosin Motor Share Functional Defects
Abstract
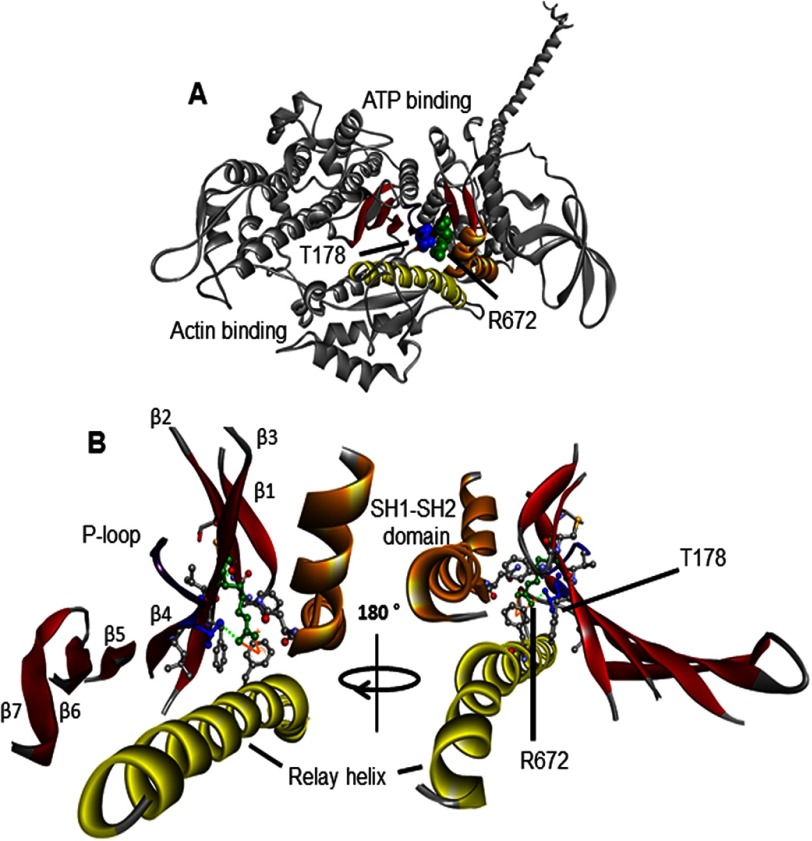
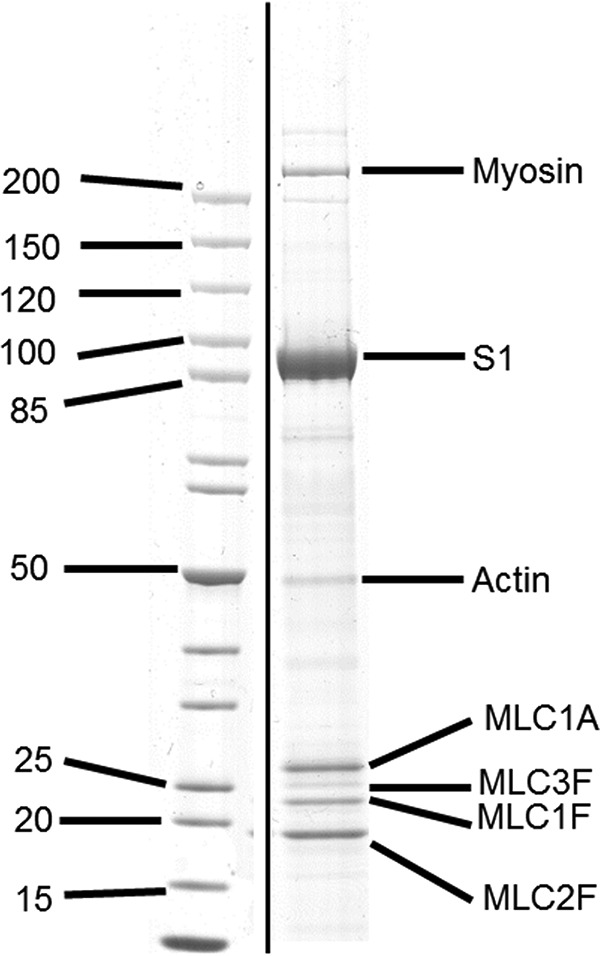
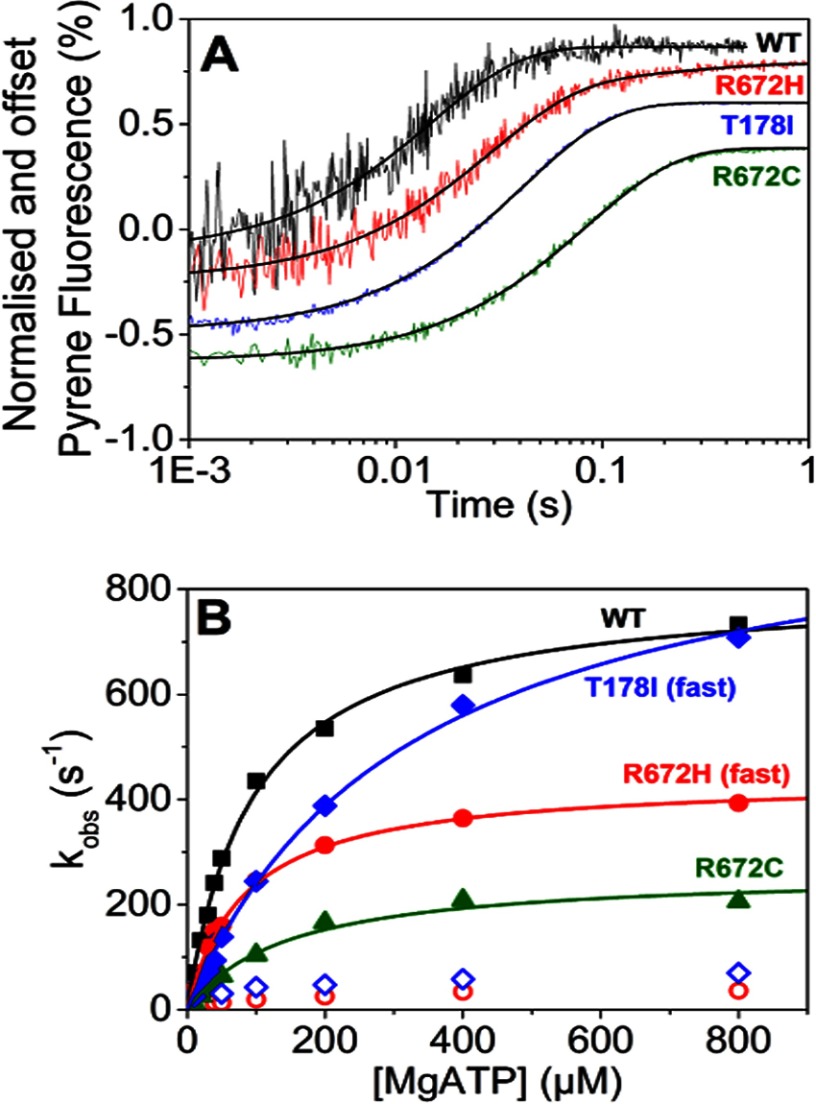
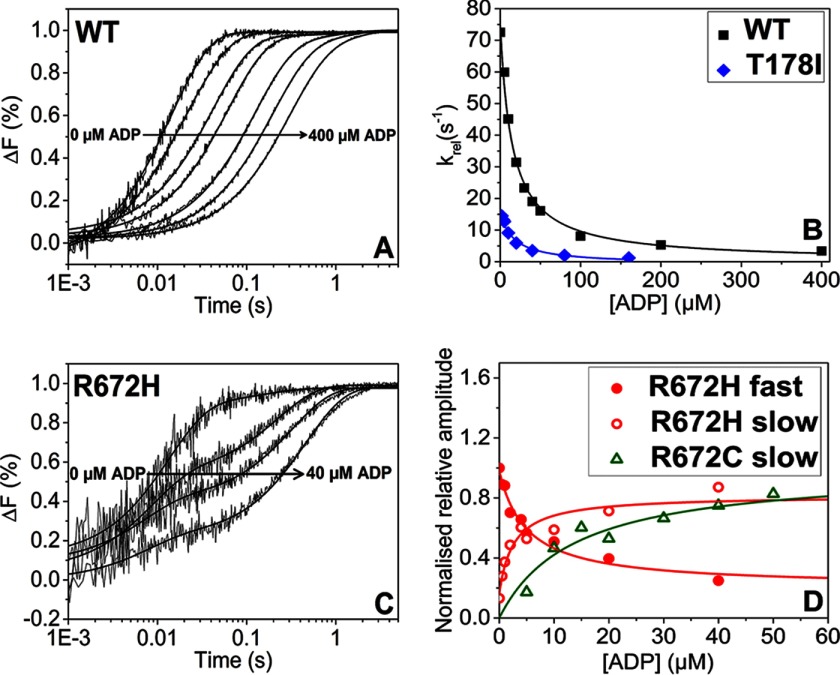
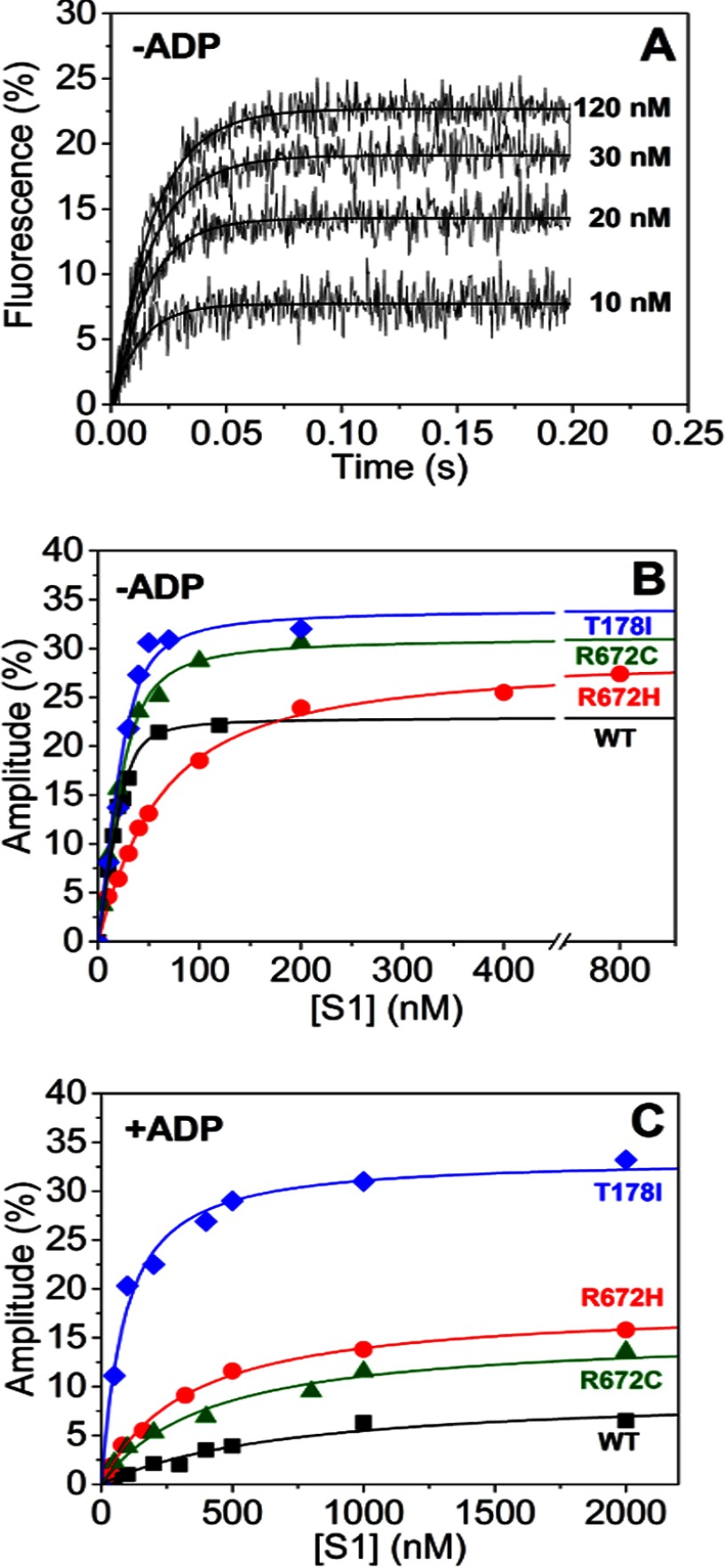
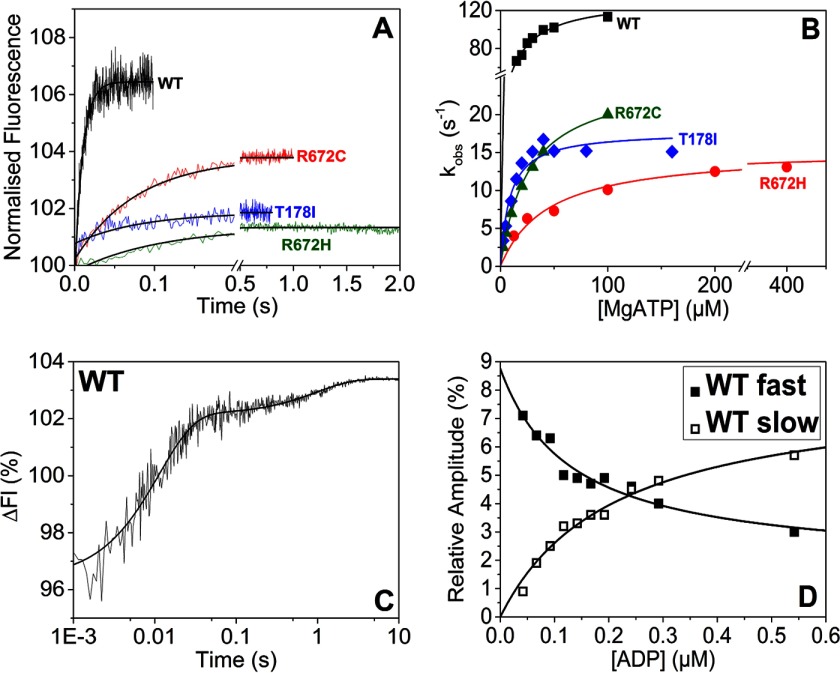
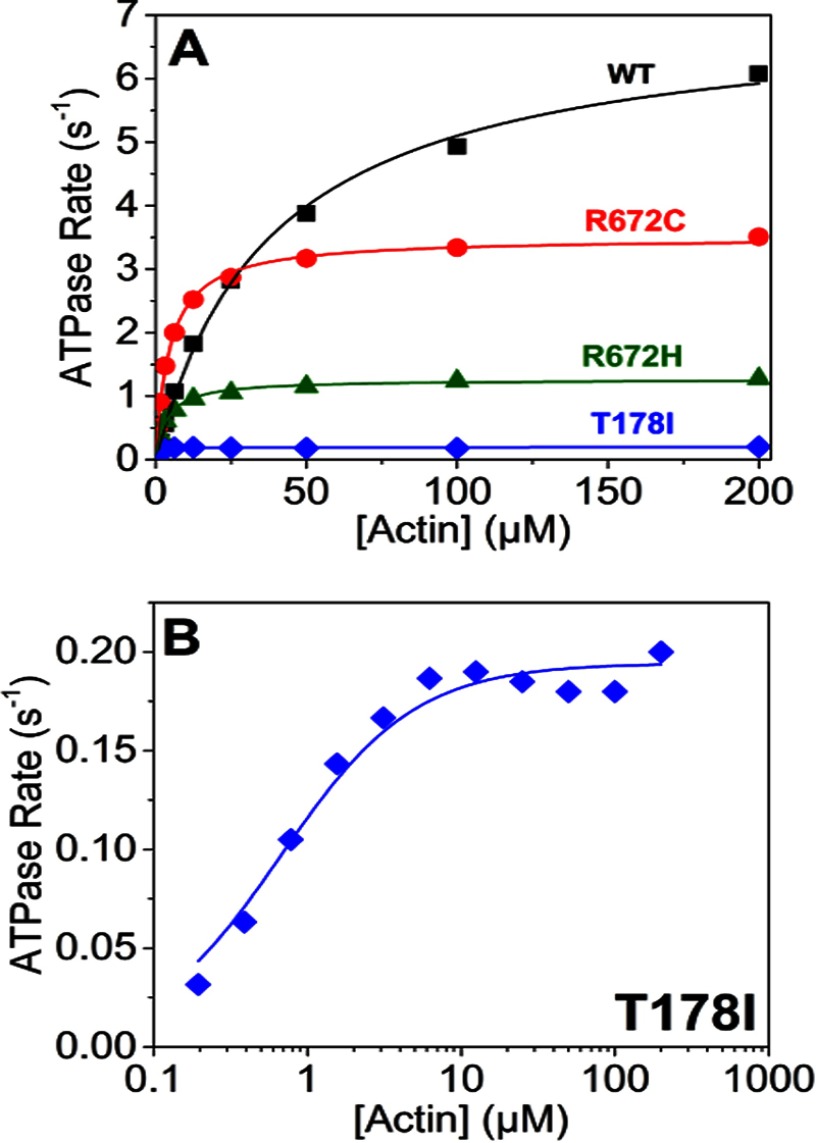
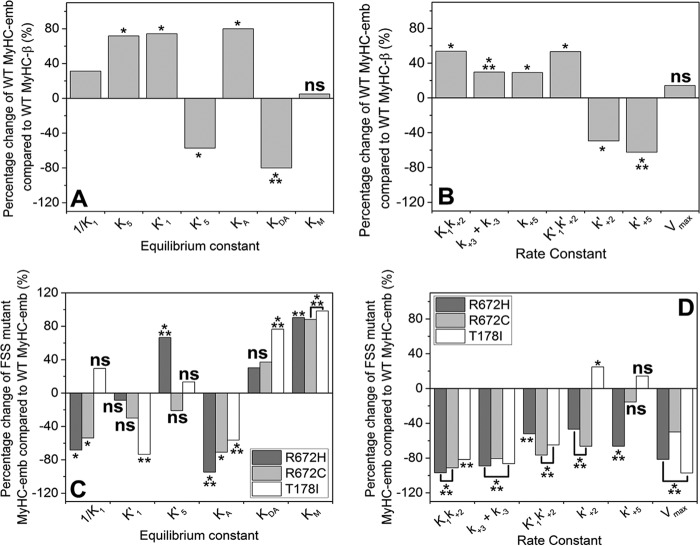
The embryonic myosin isoform is expressed during fetal development and rapidly down-regulated after birth. Freeman-Sheldon syndrome (FSS) is a disease associated with missense mutations in the motor domain of this myosin. It is the most severe form of distal arthrogryposis, leading to overcontraction of the hands, feet, and orofacial muscles and other joints of the body. Availability of human embryonic muscle tissue has been a limiting factor in investigating the properties of this isoform and its mutations. Using a recombinant expression system, we have studied homogeneous samples of human motors for the WT and three of the most common FSS mutants: R672H, R672C, and T178I. Our data suggest that the WT embryonic myosin motor is similar in contractile speed to the slow type I/β cardiac based on the rate constant for ADP release and ADP affinity for actin-myosin. All three FSS mutations show dramatic changes in kinetic properties, most notably the slowing of the apparent ATP hydrolysis step (reduced 5-9-fold), leading to a longer lived detached state and a slowed Vmax of the ATPase (2-35-fold), indicating a slower cycling time. These mutations therefore seriously disrupt myosin function.
Keywords: ATPase; enzyme kinetics; human myosin; molecular motor; muscle disease; myosin subfragment 1; recombinant protein expression; skeletal muscle; transient kinetics.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
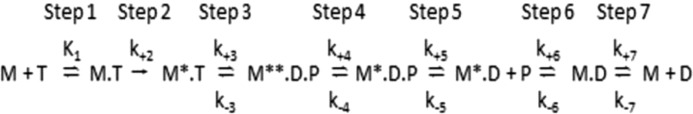
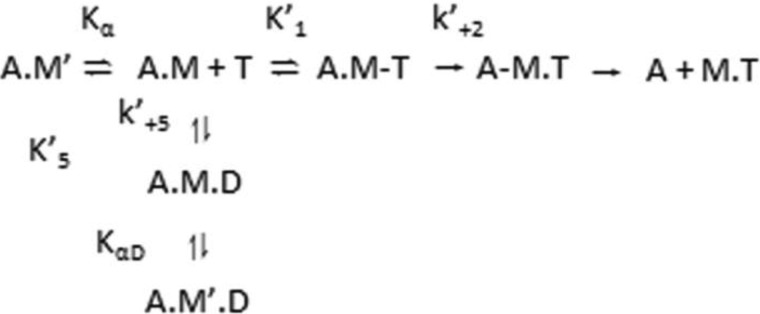
References
-
- Oldfors A. (2007) Hereditary myosin myopathies. Neuromusc. Disord. 17, 355–367 - PubMed
-
- Clarke N. F., Amburgey K., Teener J., Camelo-Piragua S., Kesari A., Punetha J., Waddell L. B., Davis M., Laing N. G., Monnier N., North K. N., Hoffman E. P., and Dowling J. J. (2013) A novel mutation expands the genetic and clinical spectrum of MYH7-related myopathies. Neuromusc. Disord. 23, 432–436 - PMC - PubMed
-
- Lamont P. J., Wallefeld W., Hilton-Jones D., Udd B., Argov Z., Barboi A.C., Bonneman C., Boycott K. M., Bushby K., Connolly A. M., Davies N., Beggs A. H., Cox G. F., Dastgir J., DeChene E. T., et al. (2014) Novel mutations widen the phenotypic spectrum of slow skeletal/β-cadiac myosin (MYH7) distal myopathy. Human Mutation 35, 868–879 - PMC - PubMed
-
- Yüceyar N., Ayhan Ö., Karasoy H., and Tolun A. (2015) Homozygous MYH7R1820W mutation results in recessive myosin storage myopathy: scapuloperoneal and respiratory weakness with dilated cardiomyopathy. Neuromusc. Disord. 25, 340–344 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
