

Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics
- PMID: 26945082
- PMCID: PMC4888539
- DOI: 10.1152/ajpheart.00858.2015
Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics
Abstract
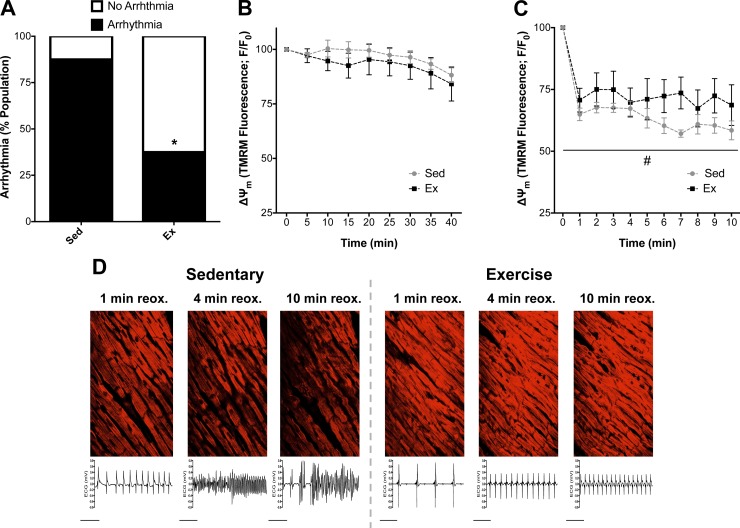
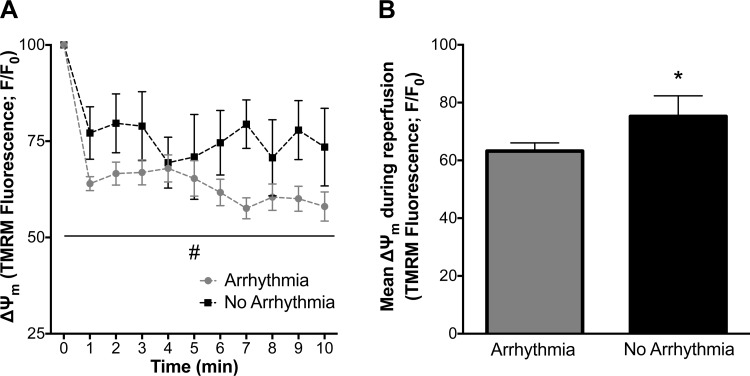
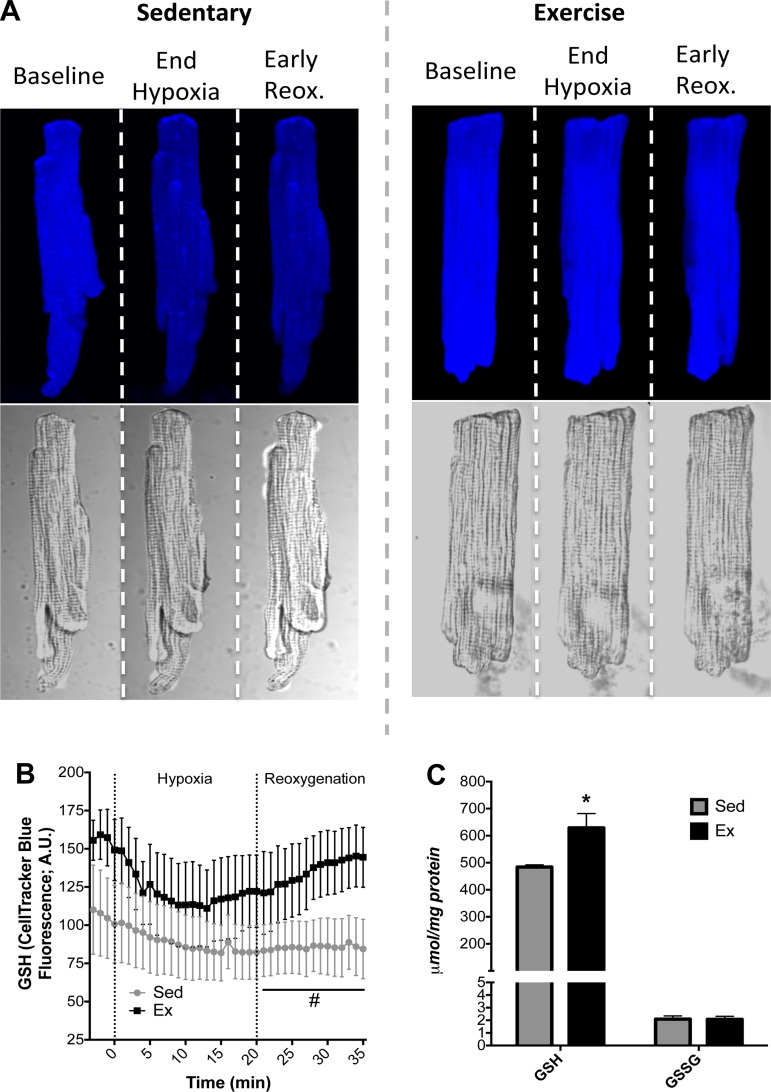
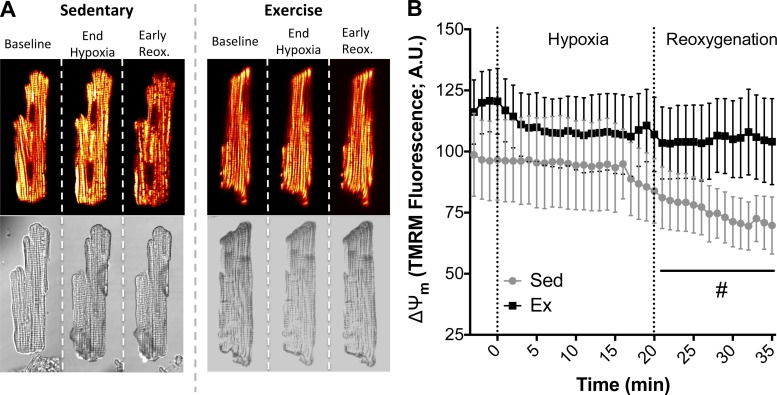
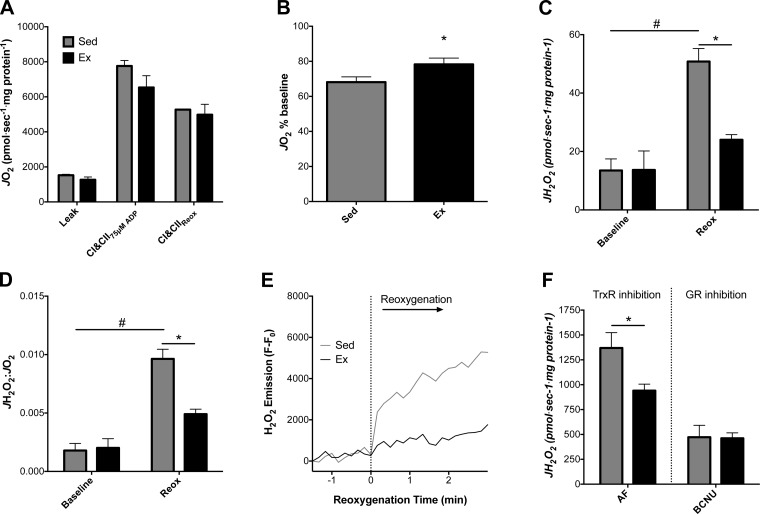
Mitochondria influence cardiac electrophysiology through energy- and redox-sensitive ion channels in the sarcolemma, with the collapse of energetics believed to be centrally involved in arrhythmogenesis. This study was conducted to determine if preservation of mitochondrial membrane potential (ΔΨm) contributes to the antiarrhythmic effect of exercise. We utilized perfused hearts, isolated myocytes, and isolated mitochondria exposed to metabolic challenge to determine the effects of exercise on cardiac mitochondria. Hearts from sedentary (Sed) and exercised (Ex; 10 days of treadmill running) Sprague-Dawley rats were perfused on a two-photon microscope stage for simultaneous measurement of ΔΨm and ECG. After ischemia-reperfusion, the collapse of ΔΨm was commensurate with the onset of arrhythmia. Exercise preserved ΔΨm and decreased the incidence of fibrillation/tachycardia (P < 0.05). Our findings in intact hearts were corroborated in isolated myocytes exposed to in vitro hypoxia-reoxygenation, with Ex rats demonstrating enhanced redox control and sustained ΔΨm during reoxygenation. Finally, we induced anoxia-reoxygenation in isolated mitochondria using high-resolution respirometry with simultaneous measurement of respiration and H2O2 Mitochondria from Ex rats sustained respiration with lower rates of H2O2 emission than Sed rats. Exercise helps sustain postischemic mitochondrial bioenergetics and redox homeostasis, which is associated with preserved ΔΨm and protection against reperfusion arrhythmia. The reduction of fatal ventricular arrhythmias through exercise-induced mitochondrial adaptations indicates that mitochondrial therapeutics may be an effective target for the treatment of heart disease.
Keywords: arrhythmia; cardioprotection; exercise; membrane potential; mitochondria.
Copyright © 2016 the American Physiological Society.
Figures
References
-
- Aiello EA, Jabr RI, Cole WC. Arrhythmia and delayed recovery of cardiac action potential during reperfusion after ischemia. Role of oxygen radical-induced no-reflow phenomenon. Circ Res 77: 153–162, 1995. - PubMed
-
- Akita Y, Otani H, Matsuhisa S, Kyoi S, Enoki C, Hattori R, Imamura H, Kamihata H, Kimura Y, Iwasaka T. Exercise-induced activation of cardiac sympathetic nerve triggers cardioprotection via redox-sensitive activation of eNOS and upregulation of iNOS. Am J Physiol Heart Circ Physiol 292: H2051–H2059, 2007. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical