

Original Research: Generation of non-deletional hereditary persistence of fetal hemoglobin β-globin locus yeast artificial chromosome transgenic mouse models: -175 Black HPFH and -195 Brazilian HPFH
- PMID: 26946532
- PMCID: PMC4871743
- DOI: 10.1177/1535370216636724
Original Research: Generation of non-deletional hereditary persistence of fetal hemoglobin β-globin locus yeast artificial chromosome transgenic mouse models: -175 Black HPFH and -195 Brazilian HPFH
Abstract
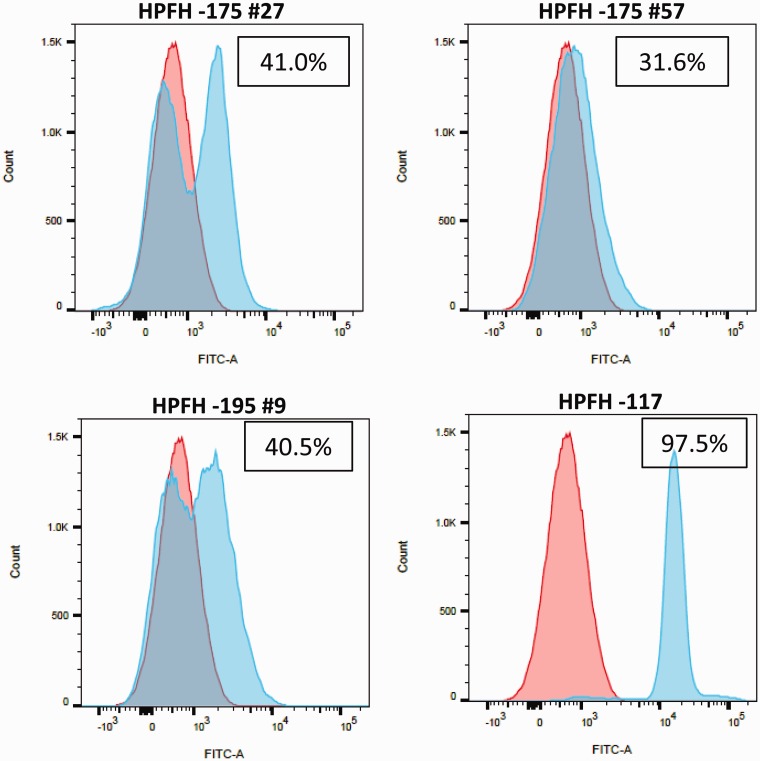
Fetal hemoglobin is a major genetic modifier of the phenotypic heterogeneity in patients with sickle cell disease and certain β-thalassemias. Normal levels of fetal hemoglobin postnatally are approximately 1% of total hemoglobin. Patients who have hereditary persistence of fetal hemoglobin, characterized by elevated synthesis of γ-globin in adulthood, show reduced disease pathophysiology. Hereditary persistence of fetal hemoglobin is caused by β-globin locus deletions (deletional hereditary persistence of fetal hemoglobin) or γ-globin gene promoter point mutations (non-deletional hereditary persistence of fetal hemoglobin). Current research has focused on elucidating the pathways involved in the maintenance/reactivation of γ-globin in adult life. To better understand these pathways, we generated new β-globin locus yeast artificial chromosome transgenic mice bearing the (A)γ-globin -175 T > C or -195 C > G hereditary persistence of fetal hemoglobin mutations to model naturally occurring hereditary persistence of fetal hemoglobin. Adult -175 and -195 mutant β-YAC mice displayed a hereditary persistence of fetal hemoglobin phenotype, as measured at the mRNA and protein levels. The molecular basis for these phenotypes was examined by chromatin immunoprecipitation of transcription factor/co-factor binding, including YY1, PAX1, TAL1, LMO2, and LDB1. In -175 HPFH versus wild-type samples, the occupancy of LMO2, TAL1 and LDB1 proteins was enriched in HPFH mice (5.8-fold, 5.2-fold and 2.7-fold, respectively), a result that concurs with a recent study in cell lines showing that these proteins form a complex with GATA-1 to mediate long-range interactions between the locus control region and the (A)γ-globin gene. Both hereditary persistence of fetal hemoglobin mutations result in a gain of (A)γ-globin activation, in contrast to other hereditary persistence of fetal hemoglobin mutations that result in a loss of repression. The mice provide additional tools to study γ-globin gene expression and may reveal new targets for selectively activating fetal hemoglobin.
Keywords: Globin gene; HPFH; fetal hemoglobin; hemoglobinopathies; sickle cell disease; transgenic mice.
© 2016 by the Society for Experimental Biology and Medicine.
Figures



Similar articles
-
{gamma}-Globin gene expression in chemical inducer of dimerization (CID)-dependent multipotential cells established from human {beta}-globin locus yeast artificial chromosome ({beta}-YAC) transgenic mice.J Biol Chem. 2005 Nov 4;280(44):36642-7. doi: 10.1074/jbc.M504402200. Epub 2005 Aug 30. J Biol Chem. 2005. PMID: 16131492
-
Mi2β is required for γ-globin gene silencing: temporal assembly of a GATA-1-FOG-1-Mi2 repressor complex in β-YAC transgenic mice.PLoS Genet. 2012;8(12):e1003155. doi: 10.1371/journal.pgen.1003155. Epub 2012 Dec 20. PLoS Genet. 2012. PMID: 23284307 Free PMC article.
-
Use of yeast artificial chromosomes (YACs) in studies of mammalian development: production of beta-globin locus YAC mice carrying human globin developmental mutants.Proc Natl Acad Sci U S A. 1995 Jun 6;92(12):5655-9. doi: 10.1073/pnas.92.12.5655. Proc Natl Acad Sci U S A. 1995. PMID: 7539923 Free PMC article.
-
Molecular basis of hereditary persistence of fetal hemoglobin.Ann N Y Acad Sci. 1998 Jun 30;850:38-44. doi: 10.1111/j.1749-6632.1998.tb10460.x. Ann N Y Acad Sci. 1998. PMID: 9668525 Review.
-
DNA sequences regulating human globin gene transcription in nondeletional hereditary persistence of fetal hemoglobin.Hemoglobin. 1989;13(6):523-41. doi: 10.3109/03630268908993104. Hemoglobin. 1989. PMID: 2481658 Review.
Cited by
-
CRISPR technology in human diseases.MedComm (2020). 2024 Jul 29;5(8):e672. doi: 10.1002/mco2.672. eCollection 2024 Aug. MedComm (2020). 2024. PMID: 39081515 Free PMC article. Review.
-
Sickle cell disease severity: an introduction.Exp Biol Med (Maywood). 2016 Apr;241(7):677-8. doi: 10.1177/1535370216641880. Exp Biol Med (Maywood). 2016. PMID: 27190296 Free PMC article. No abstract available.
-
Reactivation of γ-globin in adult β-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing.Blood. 2018 Jun 28;131(26):2915-2928. doi: 10.1182/blood-2018-03-838540. Epub 2018 May 22. Blood. 2018. PMID: 29789357 Free PMC article.
-
Rapid and Sensitive Assessment of Globin Chains for Gene and Cell Therapy of Hemoglobinopathies.Hum Gene Ther Methods. 2018 Feb;29(1):60-74. doi: 10.1089/hgtb.2017.190. Hum Gene Ther Methods. 2018. PMID: 29325430 Free PMC article.
-
Biomarkers of clinical severity in treated and untreated sickle cell disease: a comparison by genotypes of a single center cohort and African Americans in the NHANES study.Br J Haematol. 2021 Aug;194(4):767-778. doi: 10.1111/bjh.17682. Epub 2021 Jul 15. Br J Haematol. 2021. PMID: 34268729 Free PMC article.
References
-
- Amoyal I, Fibach E. Hemoglobin switch in the newborn: a flow cytometry analysis. Neonatology 2007; 91: 61–8. - PubMed
-
- Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann NY Acad Sci 1998; 850: 39–44. - PubMed
-
- Wood WG. Hereditary persistence and fetal hemoglobin and δβthalassemia. Disorders of hemoglobin. In: Steinberg MH, Forget BG, Higgs D, Nagel RL. (eds). Disorders of hemoglobin, Cambridge, UK: Cambridge University Press, 2001.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources