

Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases
- PMID: 26950747
- PMCID: PMC4967716
- DOI: 10.1038/nmeth.3799
Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases
Abstract
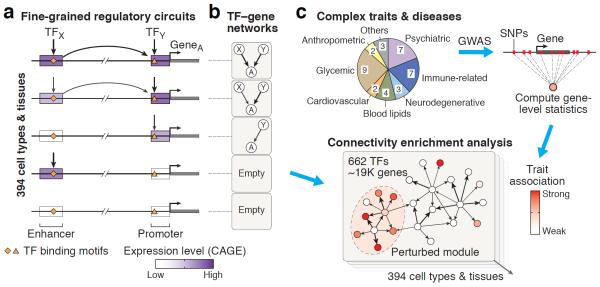
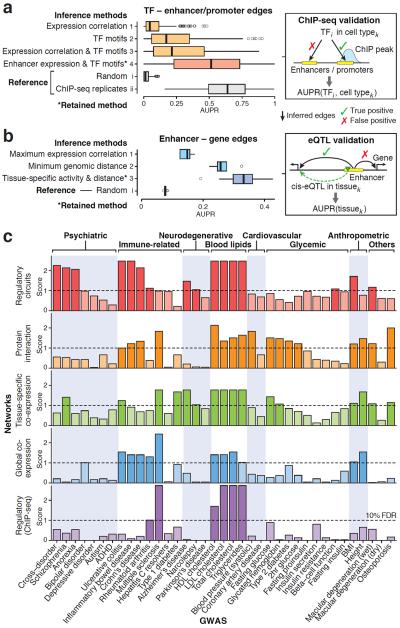
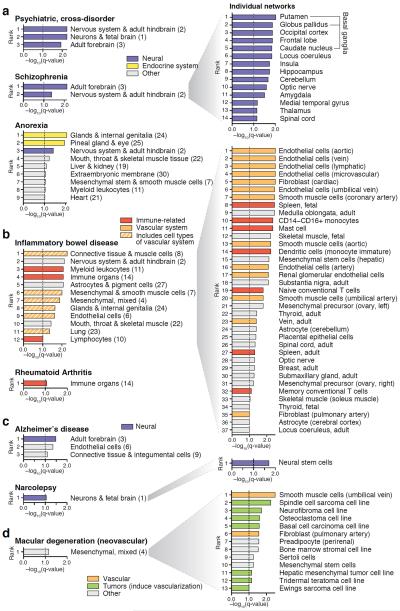
Mapping perturbed molecular circuits that underlie complex diseases remains a great challenge. We developed a comprehensive resource of 394 cell type- and tissue-specific gene regulatory networks for human, each specifying the genome-wide connectivity among transcription factors, enhancers, promoters and genes. Integration with 37 genome-wide association studies (GWASs) showed that disease-associated genetic variants--including variants that do not reach genome-wide significance--often perturb regulatory modules that are highly specific to disease-relevant cell types or tissues. Our resource opens the door to systematic analysis of regulatory programs across hundreds of human cell types and tissues (http://regulatorycircuits.org).
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
