

Prediction, dynamics, and visualization of antigenic phenotypes of seasonal influenza viruses
- PMID: 26951657
- PMCID: PMC4812706
- DOI: 10.1073/pnas.1525578113
Prediction, dynamics, and visualization of antigenic phenotypes of seasonal influenza viruses
Abstract
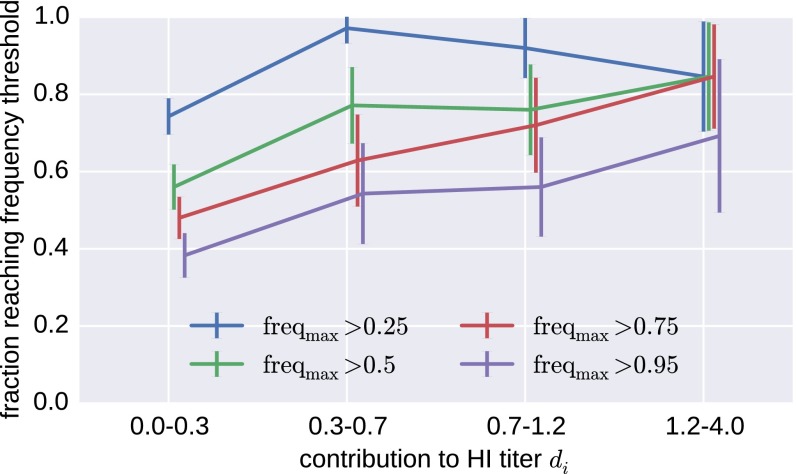
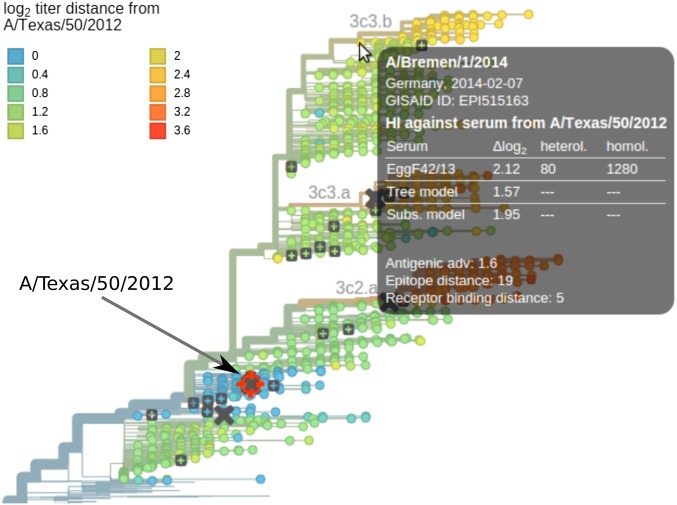
Human seasonal influenza viruses evolve rapidly, enabling the virus population to evade immunity and reinfect previously infected individuals. Antigenic properties are largely determined by the surface glycoprotein hemagglutinin (HA), and amino acid substitutions at exposed epitope sites in HA mediate loss of recognition by antibodies. Here, we show that antigenic differences measured through serological assay data are well described by a sum of antigenic changes along the path connecting viruses in a phylogenetic tree. This mapping onto the tree allows prediction of antigenicity from HA sequence data alone. The mapping can further be used to make predictions about the makeup of the future A(H3N2) seasonal influenza virus population, and we compare predictions between models with serological and sequence data. To make timely model output readily available, we developed a web browser-based application that visualizes antigenic data on a continuously updated phylogeny.
Keywords: antigenic distance; evolution; phylogenetic tree.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
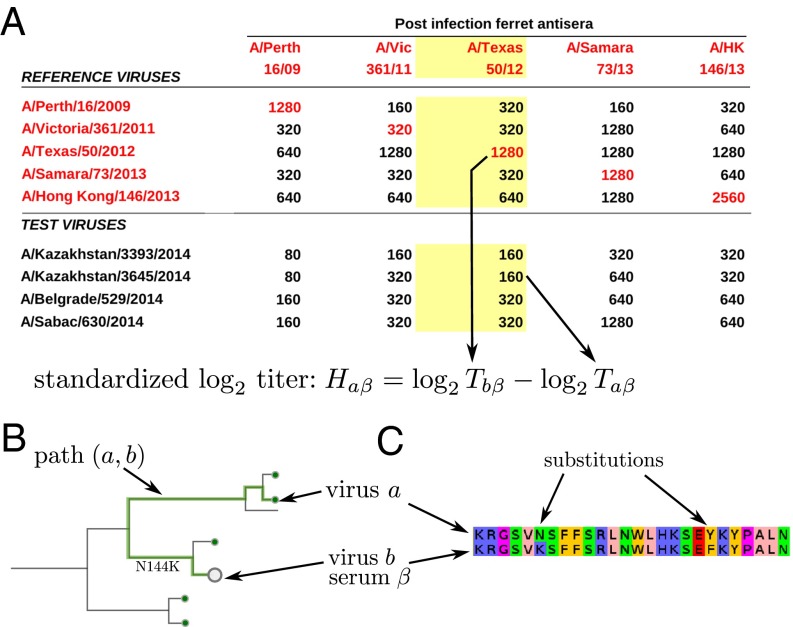
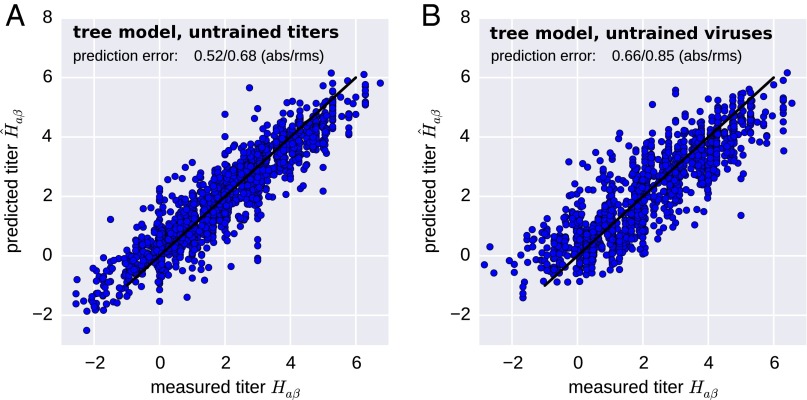
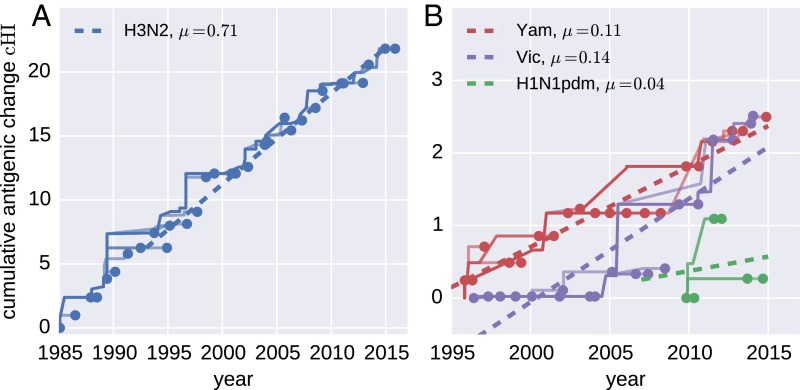
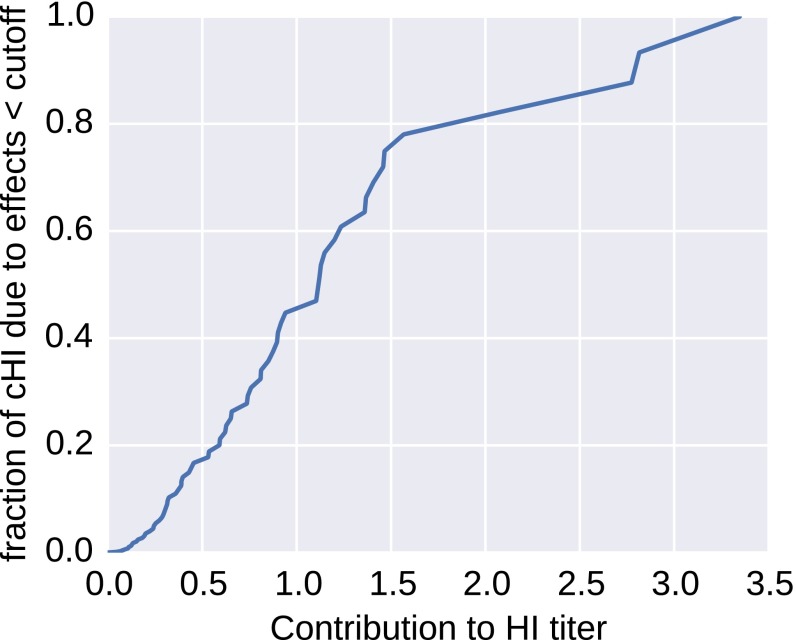
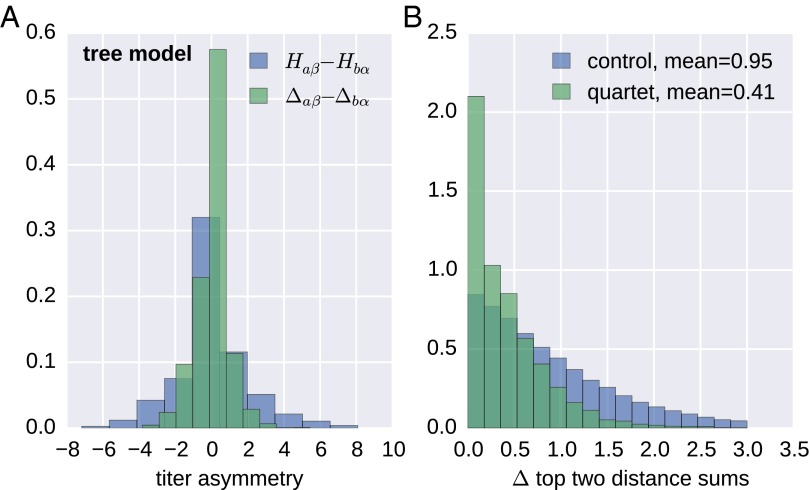
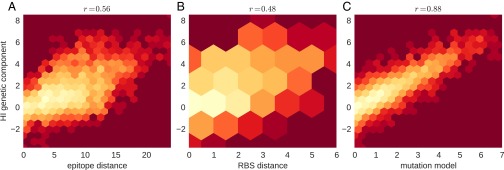
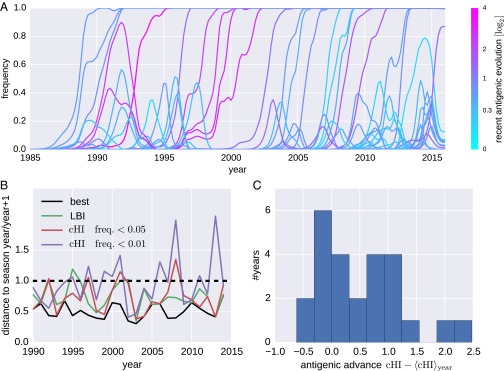
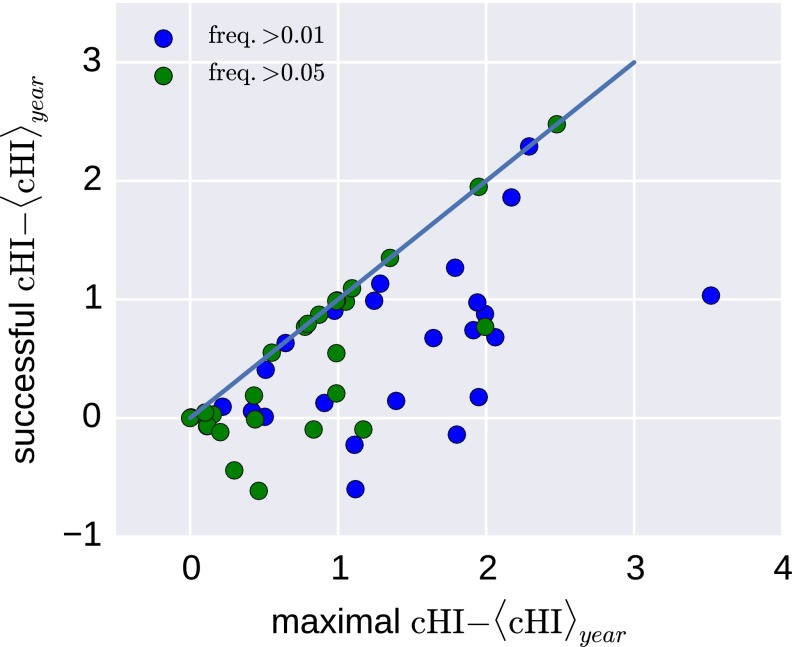
References
-
- Barr IG, et al. Writing Committee of the World Health Organization Consultation on Northern Hemisphere Influenza Vaccine Composition for 2013–2014 WHO recommendations for the viruses used in the 2013-2014 Northern Hemisphere influenza vaccine: Epidemiology, antigenic and genetic characteristics of influenza A(H1N1)pdm09, A(H3N2) and B influenza viruses collected from October 2012 to January 2013. Vaccine. 2014;32(37):4713–4725. - PubMed
-
- Smith DJ, et al. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305(5682):371–376. - PubMed
