

Nuclear DNA damage signalling to mitochondria in ageing
- PMID: 26956196
- PMCID: PMC5161407
- DOI: 10.1038/nrm.2016.14
Nuclear DNA damage signalling to mitochondria in ageing
Abstract
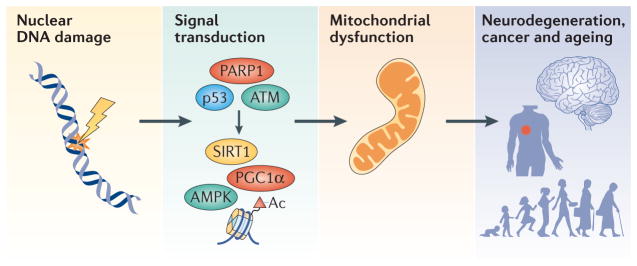
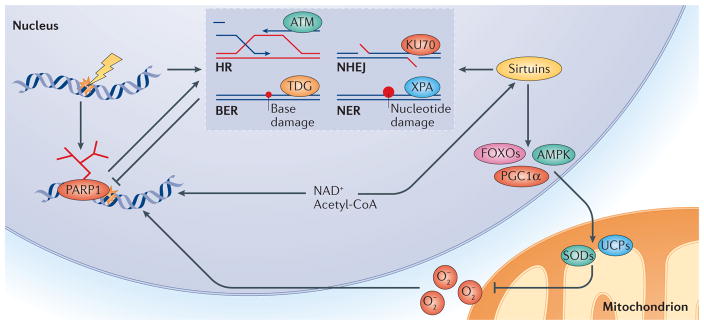
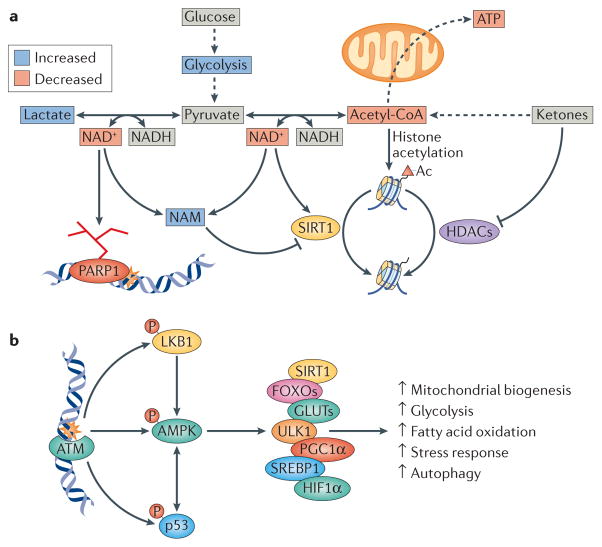
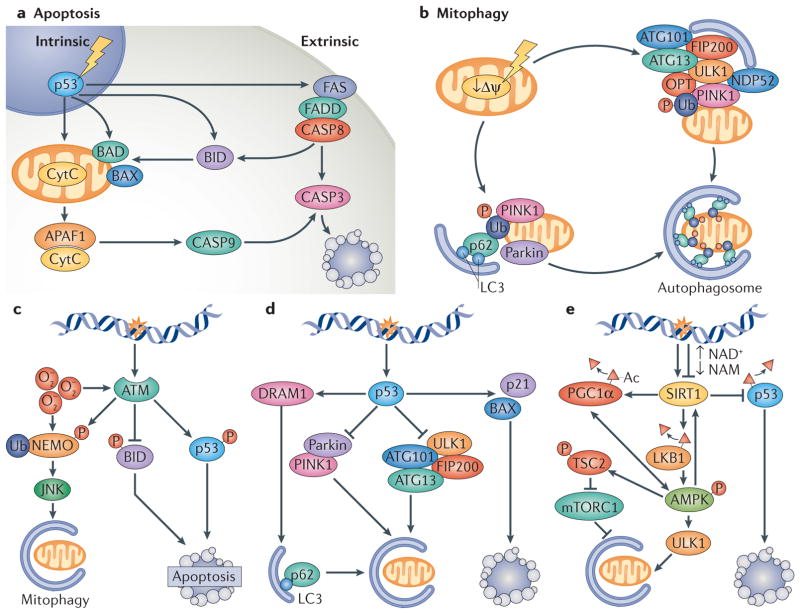
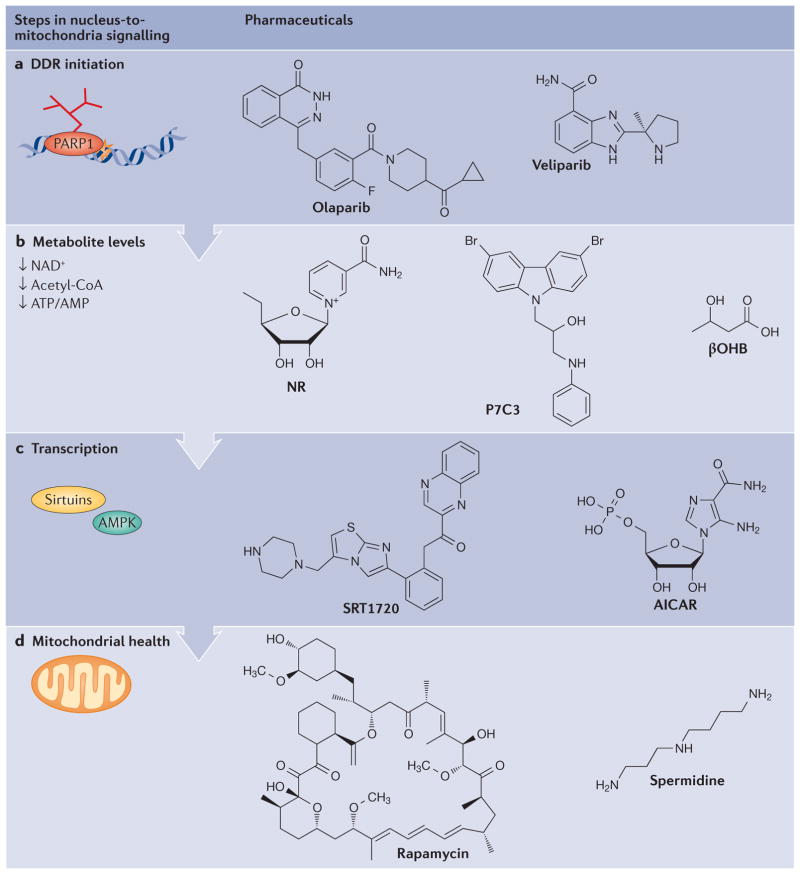
Mitochondrial dysfunction is a hallmark of ageing, and mitochondrial maintenance may lead to increased healthspan. Emerging evidence suggests a crucial role for signalling from the nucleus to mitochondria (NM signalling) in regulating mitochondrial function and ageing. An important initiator of NM signalling is nuclear DNA damage, which accumulates with age and may contribute to the development of age-associated diseases. DNA damage-dependent NM signalling constitutes a network that includes nuclear sirtuins and controls genomic stability and mitochondrial integrity. Pharmacological modulation of NM signalling is a promising novel approach for the prevention and treatment of age-associated diseases.
Conflict of interest statement
statement The authors declare competing interests: see Web version for details.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
