

CoeViz: a web-based tool for coevolution analysis of protein residues
- PMID: 26956673
- PMCID: PMC4782369
- DOI: 10.1186/s12859-016-0975-z
CoeViz: a web-based tool for coevolution analysis of protein residues
Abstract
Background: Proteins generally perform their function in a folded state. Residues forming an active site, whether it is a catalytic center or interaction interface, are frequently distant in a protein sequence. Hence, traditional sequence-based prediction methods focusing on a single residue (or a short window of residues) at a time may have difficulties in identifying and clustering the residues constituting a functional site, especially when a protein has multiple functions. Evolutionary information encoded in multiple sequence alignments is known to greatly improve sequence-based predictions. Identification of coevolving residues further advances the protein structure and function annotation by revealing cooperative pairs and higher order groupings of residues.
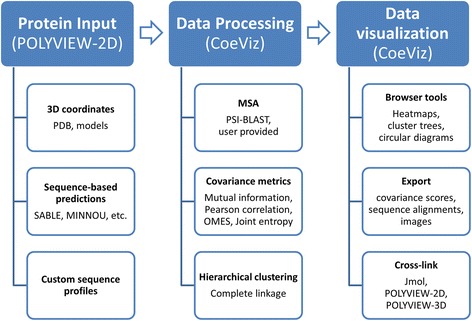
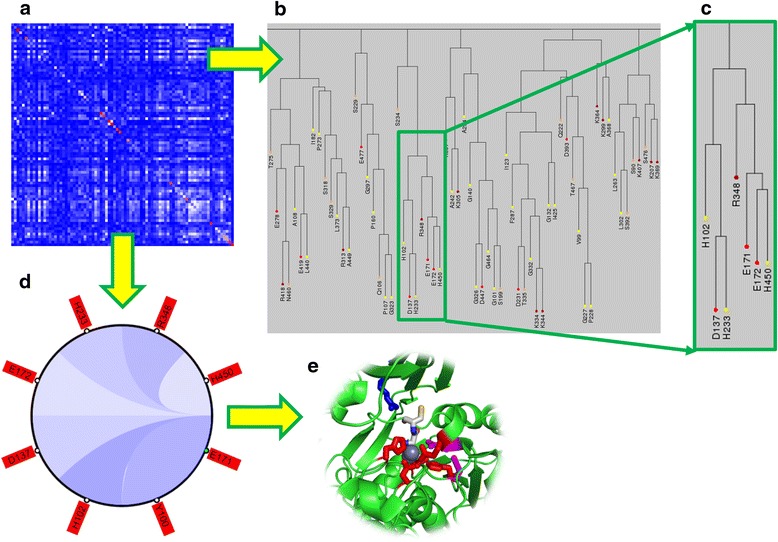
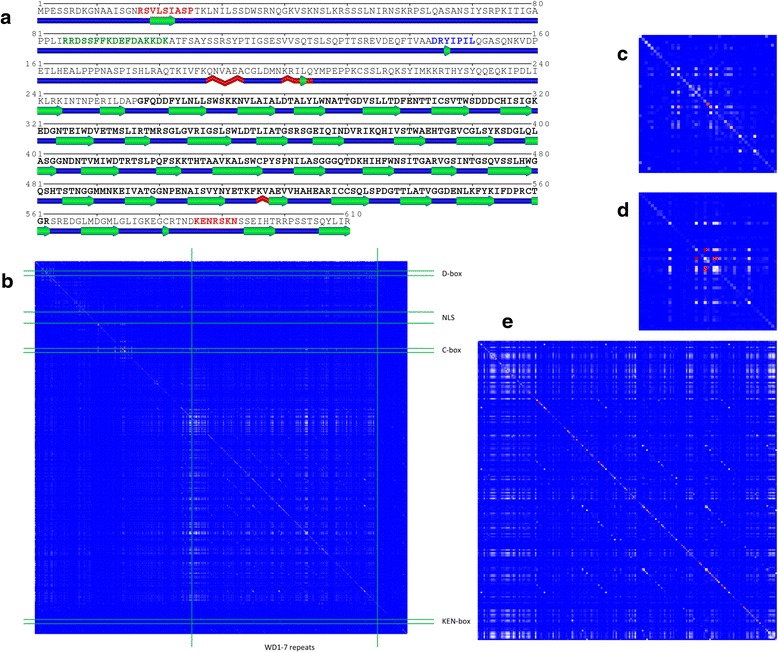
Results: We present a new web-based tool (CoeViz) that provides a versatile analysis and visualization of pairwise coevolution of amino acid residues. The tool computes three covariance metrics: mutual information, chi-square statistic, Pearson correlation, and one conservation metric: joint Shannon entropy. Implemented adjustments of covariance scores include phylogeny correction, corrections for sequence dissimilarity and alignment gaps, and the average product correction. Visualization of residue relationships is enhanced by hierarchical cluster trees, heat maps, circular diagrams, and the residue highlighting in protein sequence and 3D structure. Unlike other existing tools, CoeViz is not limited to analyzing conserved domains or protein families and can process long, unstructured and multi-domain proteins thousands of residues long. Two examples are provided to illustrate the use of the tool for identification of residues (1) involved in enzymatic function, (2) forming short linear functional motifs, and (3) constituting a structural domain.
Conclusions: CoeViz represents a practical resource for a quick sequence-based protein annotation for molecular biologists, e.g., for identifying putative functional clusters of residues and structural domains. CoeViz also can serve computational biologists as a resource of coevolution matrices, e.g., for developing machine learning-based prediction models. The presented tool is integrated in the POLYVIEW-2D server (http://polyview.cchmc.org/) and available from resulting pages of POLYVIEW-2D.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
