

Tissue-independent and tissue-specific patterns of DNA methylation alteration in cancer
- PMID: 26958079
- PMCID: PMC4782576
- DOI: 10.1186/s13072-016-0058-4
Tissue-independent and tissue-specific patterns of DNA methylation alteration in cancer
Abstract
Background: There is growing evidence that DNA methylation alterations contribute to carcinogenesis. While cancer tissue exhibits widespread DNA methylation changes, the proportion of tissue-specific versus tissue-independent DNA methylation alterations in cancer is unclear. In addition, it is unknown which factors determine the patterns of aberrant DNA methylation in cancer.
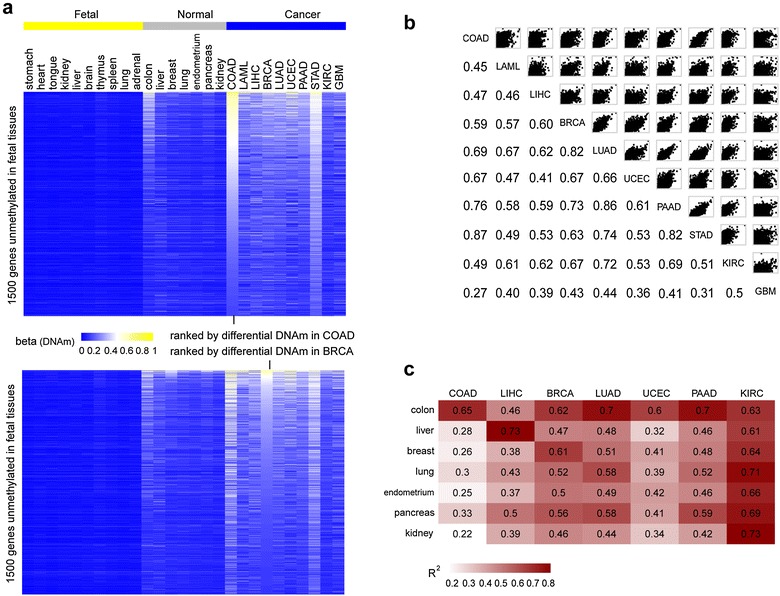
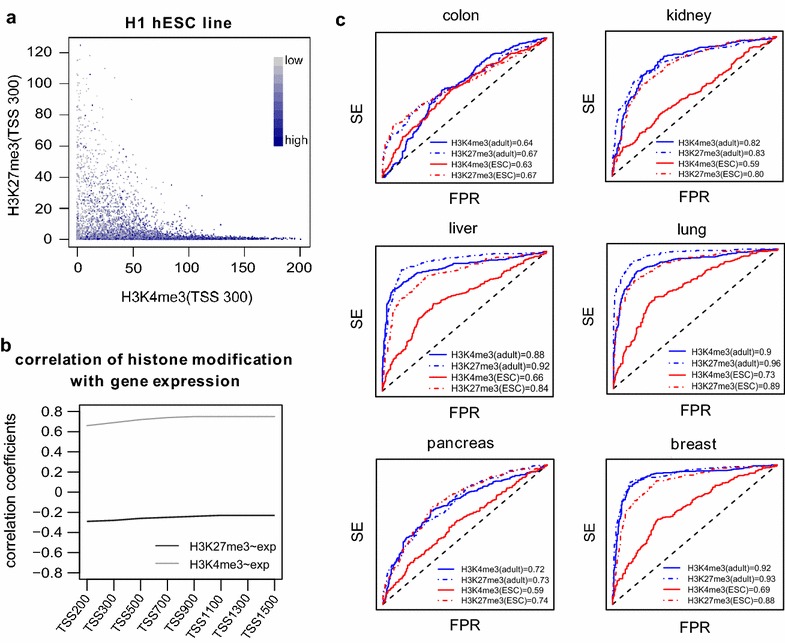
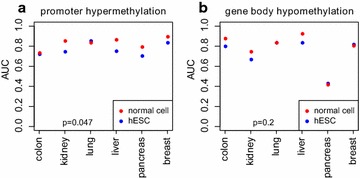
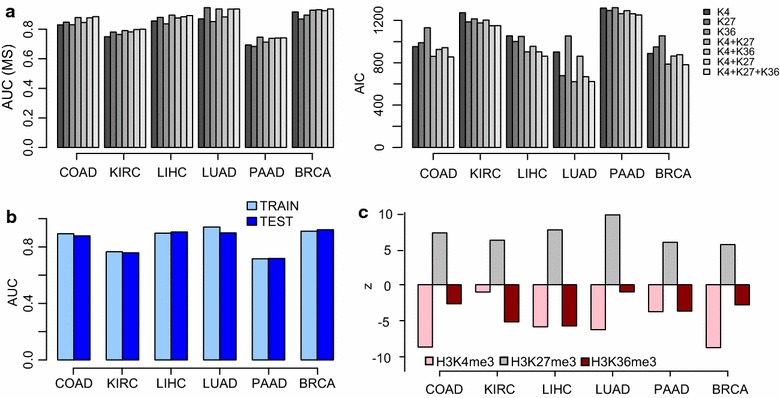
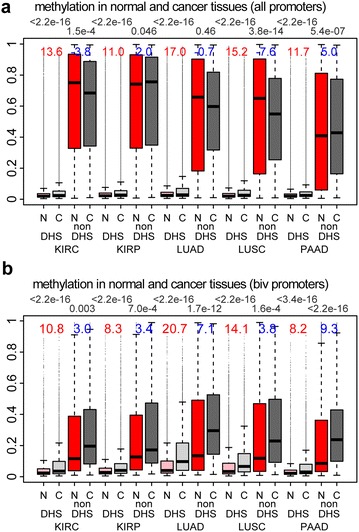
Results: Using HumanMethylation450 BeadChips (450k), we here analyze genome-wide DNA methylation patterns of ten types of fetal tissue, in addition to matched normal-cancer data for corresponding tissue types, encompassing over 3000 samples. We demonstrate that the level of aberrant cancer DNA methylation in gene promoters and gene bodies is highly correlated between cancer types. We estimate that up to 60 % of the DNA methylation variation in a cancer genome of a given tissue type is explained by the corresponding variation in a cancer genome of another type, implying that much of the cancer DNA methylation landscape is tissue independent. We further show that histone marks in normal cells are better predictors of aberrant cancer DNA methylation than the corresponding signals in human embryonic stem cells. We build predictors of cancer DNA methylation patterns and show that although inclusion of three histone marks (H3K4me3, H3K27me3 and H3K36me3) improves model accuracy, the bivalent marks are the most predictive. Finally, we show that chromatin accessibility of gene promoters in normal tissue dictates the promoter's propensity to acquire aberrant DNA methylation in cancer in so far as it determines its level of DNA methylation in normal tissue.
Conclusions: Our data show that a considerable fraction of the aberrant cancer DNA methylation landscape results from a mechanism that is largely tissue specific. Histone marks as specified in the normal cell of origin provide highly predictive models of aberrant cancer DNA methylation and outperform those derived from the same marks in hESCs.
Keywords: Bivalency; Cancer; Chromatin; DNA methylation; Histone.
Figures
References
-
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2012;44:40–46. doi: 10.1038/ng.969. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
