

Multiplex methods provide effective integration of multi-omic data in genome-scale models
- PMID: 26961692
- PMCID: PMC4896256
- DOI: 10.1186/s12859-016-0912-1
Multiplex methods provide effective integration of multi-omic data in genome-scale models
Abstract
Background: Genomic, transcriptomic, and metabolic variations shape the complex adaptation landscape of bacteria to varying environmental conditions. Elucidating the genotype-phenotype relation paves the way for the prediction of such effects, but methods for characterizing the relationship between multiple environmental factors are still lacking. Here, we tackle the problem of extracting network-level information from collections of environmental conditions, by integrating the multiple omic levels at which the bacterial response is measured.
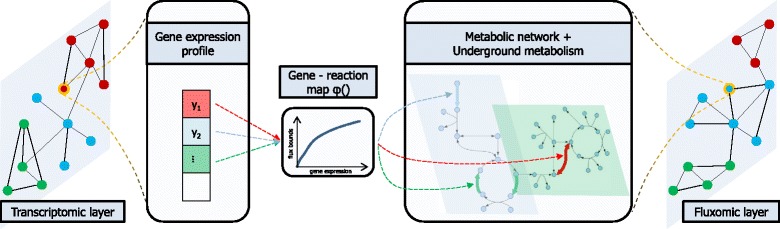
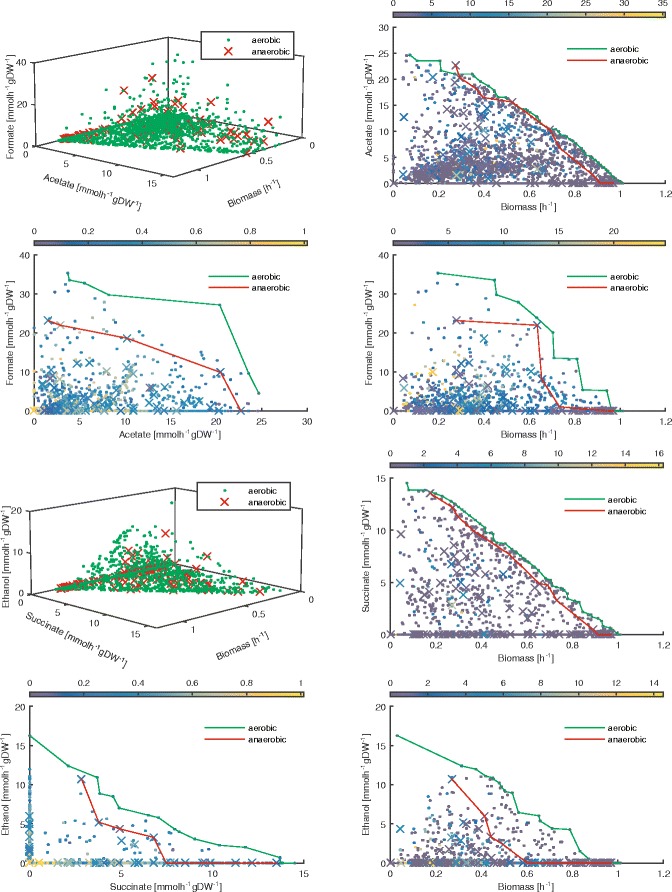
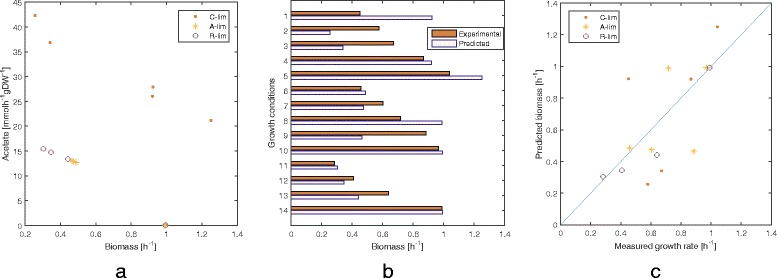
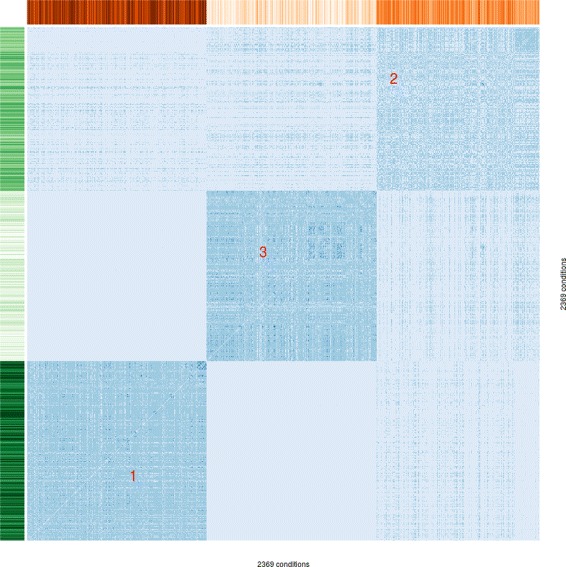
Results: To this end, we model a large compendium of growth conditions as a multiplex network consisting of transcriptomic and fluxomic layers, and we propose a multi-omic network approach to infer similarity of growth conditions by integrating layers of the multiplex network. Each node of the network represents a single condition, while edges are similarities between conditions, as measured by phenotypic and transcriptomic properties on different layers of the network. We then fuse these layers into one network, therefore capturing a global network of conditions and the associated similarities across two omic levels. We apply this multi-omic fusion to an updated genome-scale reconstruction of Escherichia coli that includes underground metabolism and new gene-protein-reaction associations.
Conclusions: Our method can be readily used to evaluate and cross-compare different collections of conditions among different species. Acquiring multi-omic information on the topology of the space of experimental conditions makes it possible to infer the position and to build condition-specific models of untested or incomplete profiles for which experimental data is not available. Our weighted network fusion method for genome-scale models is freely available at https://github.com/maxconway/SNFtool .
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
