

Genome-Wide Investigation Using sRNA-Seq, Degradome-Seq and Transcriptome-Seq Reveals Regulatory Networks of microRNAs and Their Target Genes in Soybean during Soybean mosaic virus Infection
- PMID: 26963095
- PMCID: PMC4786119
- DOI: 10.1371/journal.pone.0150582
Genome-Wide Investigation Using sRNA-Seq, Degradome-Seq and Transcriptome-Seq Reveals Regulatory Networks of microRNAs and Their Target Genes in Soybean during Soybean mosaic virus Infection
Erratum in
-
Correction: Genome-Wide Investigation Using sRNA-Seq, Degradome-Seq and Transcriptome-Seq Reveals Regulatory Networks of microRNAs and Their Target Genes in Soybean during Soybean mosaic virus Infection.PLoS One. 2017 Nov 9;12(11):e0188140. doi: 10.1371/journal.pone.0188140. eCollection 2017. PLoS One. 2017. PMID: 29121117 Free PMC article.
Abstract
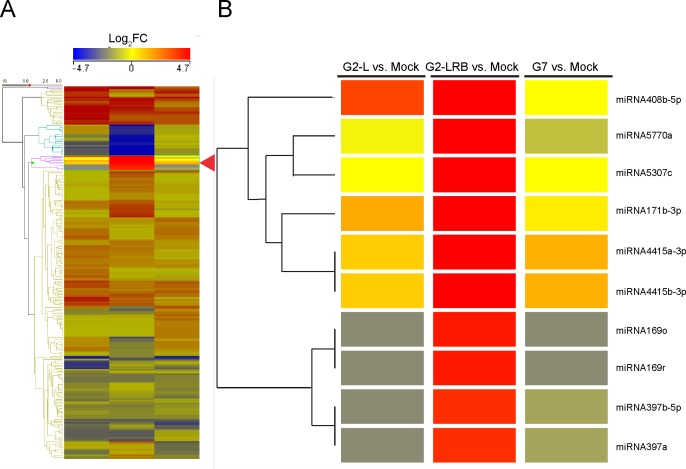
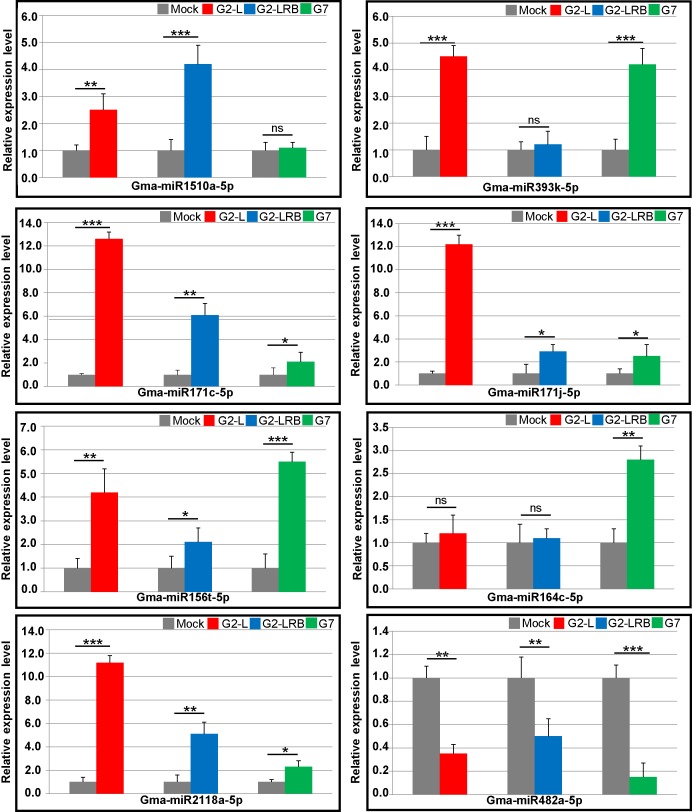
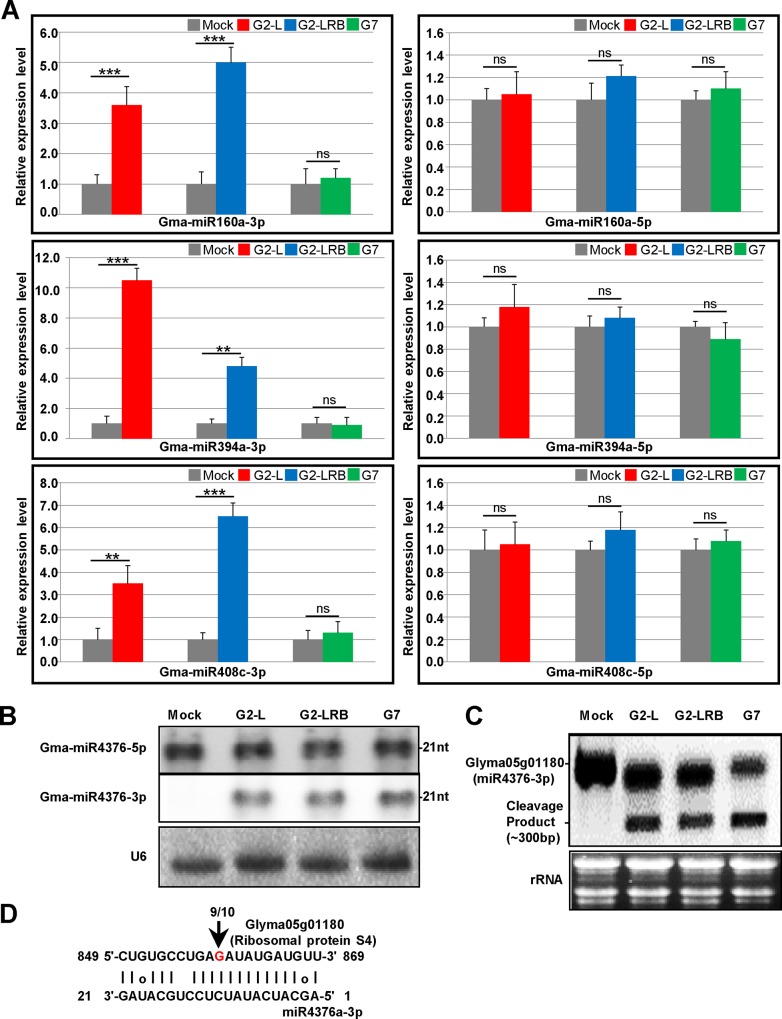
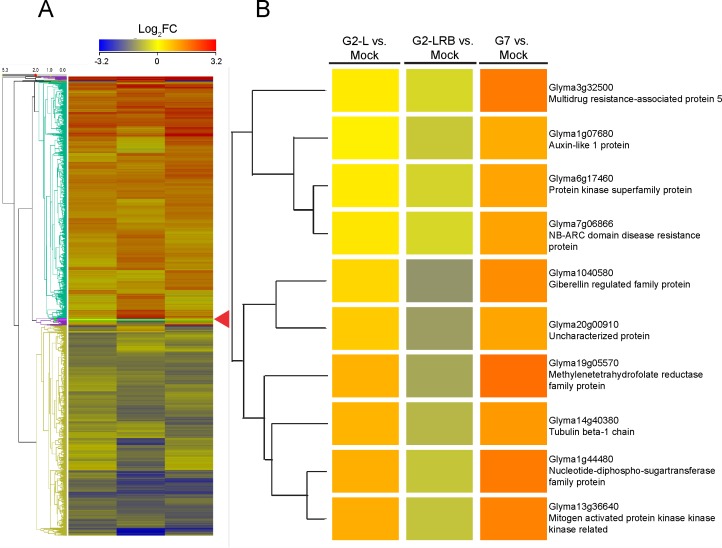
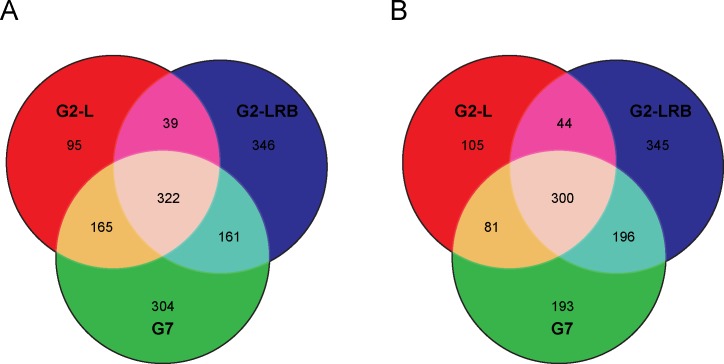
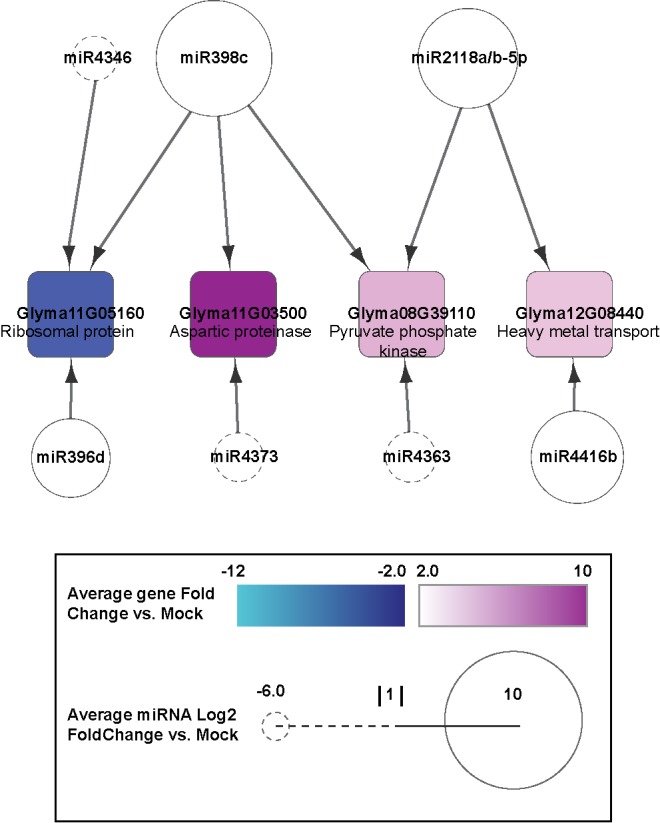
MicroRNAs (miRNAs) play key roles in a variety of cellular processes through regulation of their target gene expression. Accumulated experimental evidence has demonstrated that infections by viruses are associated with the altered expression profile of miRNAs and their mRNA targets in the host. However, the regulatory network of miRNA-mRNA interactions during viral infection remains largely unknown. In this study, we performed small RNA (sRNA)-seq, degradome-seq and as well as a genome-wide transcriptome analysis to profile the global gene and miRNA expression in soybean following infections by three different Soybean mosaic virus (SMV) isolates, L (G2 strain), LRB (G2 strain) and G7 (G7 strain). sRNA-seq analyses revealed a total of 253 soybean miRNAs with a two-fold or greater change in abundance compared with the mock-inoculated control. 125 transcripts were identified as the potential cleavage targets of 105 miRNAs and validated by degradome-seq analyses. Genome-wide transcriptome analysis showed that total 2679 genes are differentially expressed in response to SMV infection including 71 genes predicted as involved in defense response. Finally, complex miRNA-mRNA regulatory networks were derived using the RNAseq, small RNAseq and degradome data. This work represents a comprehensive, global approach to examining virus-host interactions. Genes responsive to SMV infection are identified as are their potential miRNA regulators. Additionally, regulatory changes of the miRNAs themselves are described and the regulatory relationships were supported with degradome data. Taken together these data provide new insights into molecular SMV-soybean interactions and offer candidate miRNAs and their targets for further elucidation of the SMV infection process.
Conflict of interest statement
Figures



References
-
- Gómez P, Rodríguez-Hernández A, Moury B, Aranda M. Genetic resistance for the sustainable control of plant virus diseases: breeding, mechanisms and durability. Euro J Plant Pathol. 2009; 125:1−22.
-
- Cho EK, Goodman RM. Strains of soybean mosaic virus: classification based on virulence in resistant soybean cultivars. Phytopathol. 1979; 69:467−70.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
