

Systems biology of the structural proteome
- PMID: 26969117
- PMCID: PMC4787049
- DOI: 10.1186/s12918-016-0271-6
Systems biology of the structural proteome
Abstract
Background: The success of genome-scale models (GEMs) can be attributed to the high-quality, bottom-up reconstructions of metabolic, protein synthesis, and transcriptional regulatory networks on an organism-specific basis. Such reconstructions are biochemically, genetically, and genomically structured knowledge bases that can be converted into a mathematical format to enable a myriad of computational biological studies. In recent years, genome-scale reconstructions have been extended to include protein structural information, which has opened up new vistas in systems biology research and empowered applications in structural systems biology and systems pharmacology.
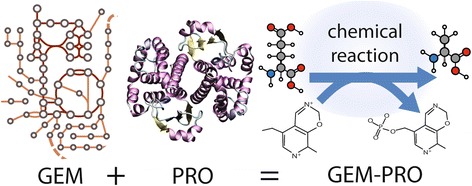
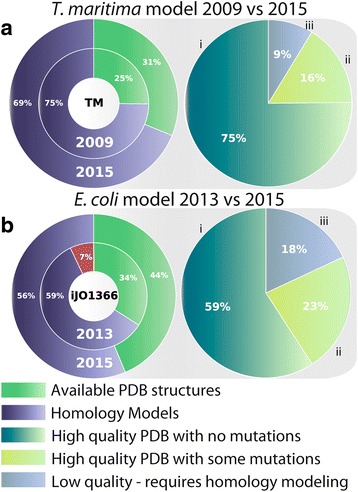
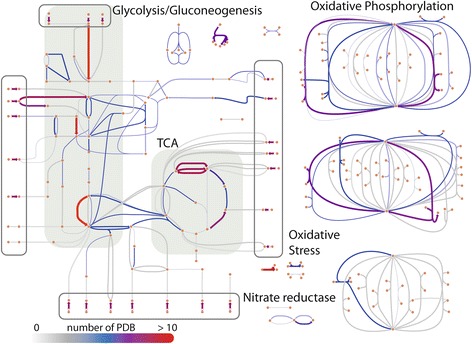
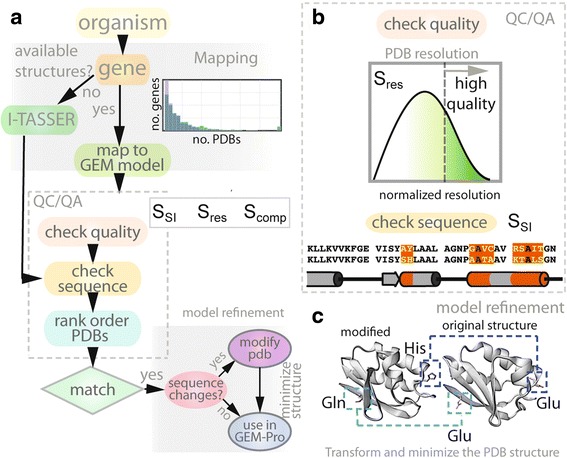
Results: Here, we present the generation, application, and dissemination of genome-scale models with protein structures (GEM-PRO) for Escherichia coli and Thermotoga maritima. We show the utility of integrating molecular scale analyses with systems biology approaches by discussing several comparative analyses on the temperature dependence of growth, the distribution of protein fold families, substrate specificity, and characteristic features of whole cell proteomes. Finally, to aid in the grand challenge of big data to knowledge, we provide several explicit tutorials of how protein-related information can be linked to genome-scale models in a public GitHub repository ( https://github.com/SBRG/GEMPro/tree/master/GEMPro_recon/).
Conclusions: Translating genome-scale, protein-related information to structured data in the format of a GEM provides a direct mapping of gene to gene-product to protein structure to biochemical reaction to network states to phenotypic function. Integration of molecular-level details of individual proteins, such as their physical, chemical, and structural properties, further expands the description of biochemical network-level properties, and can ultimately influence how to model and predict whole cell phenotypes as well as perform comparative systems biology approaches to study differences between organisms. GEM-PRO offers insight into the physical embodiment of an organism's genotype, and its use in this comparative framework enables exploration of adaptive strategies for these organisms, opening the door to many new lines of research. With these provided tools, tutorials, and background, the reader will be in a position to run GEM-PRO for their own purposes.
Figures
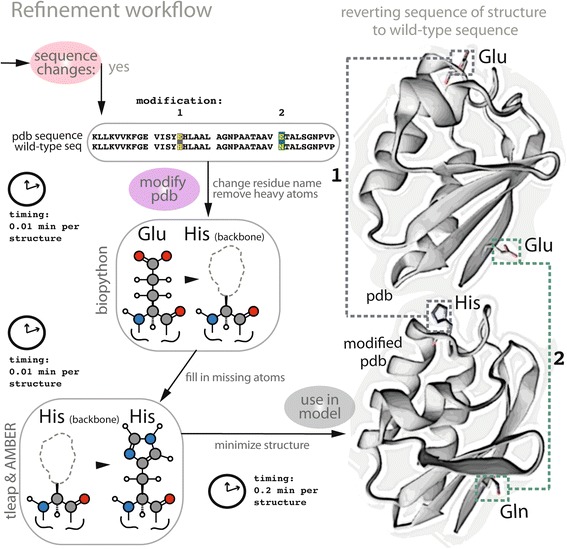
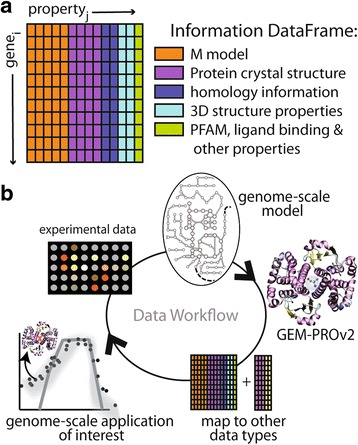
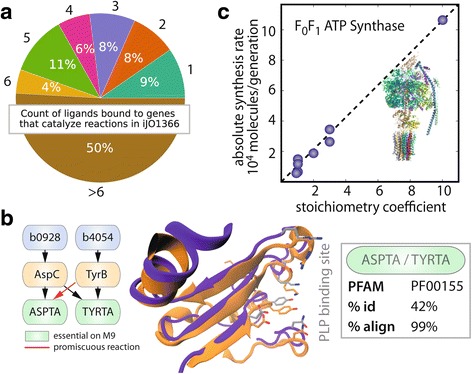
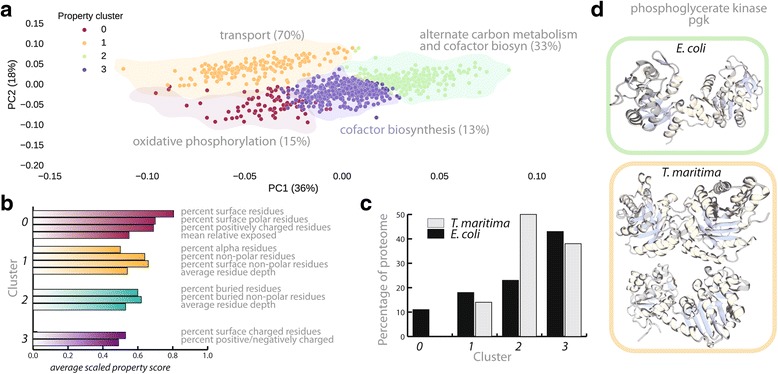
References
-
- Thiele I, Jamshidi N, Fleming RMT, Palsson BØ. Genome-scale reconstruction of Escherichia coli’s transcriptional and translational machinery: a knowledge base, its mathematical formulation, and its functional characterization. PLoS Comput Biol. 2009;5:e1000312. doi: 10.1371/journal.pcbi.1000312. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
