

Hierarchy and extremes in selections from pools of randomized proteins
- PMID: 26969726
- PMCID: PMC4822605
- DOI: 10.1073/pnas.1517813113
Hierarchy and extremes in selections from pools of randomized proteins
Abstract
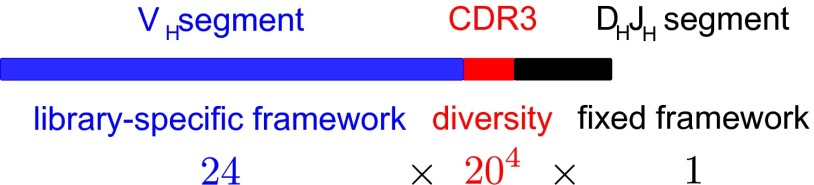
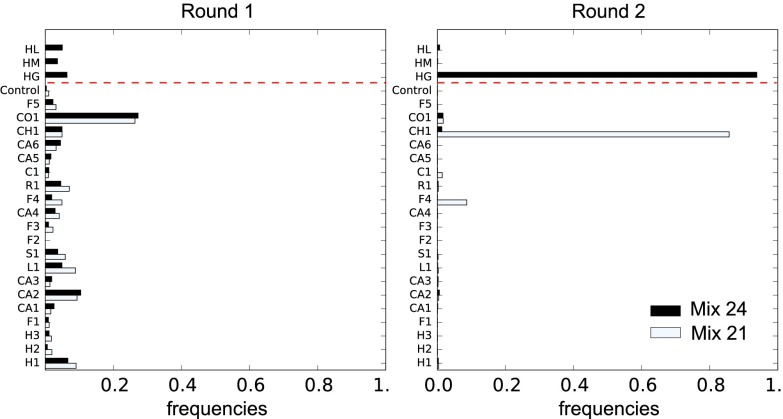
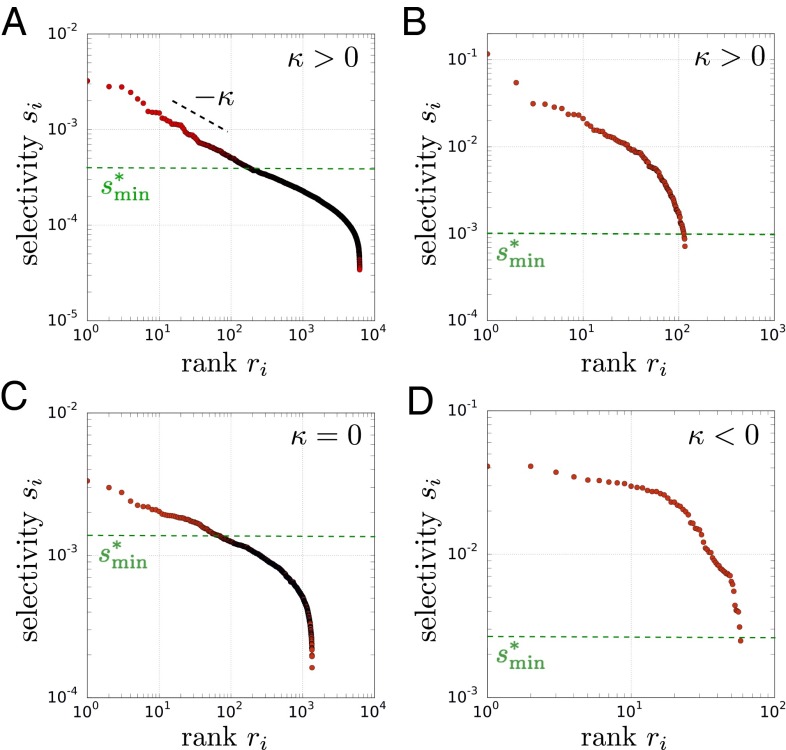
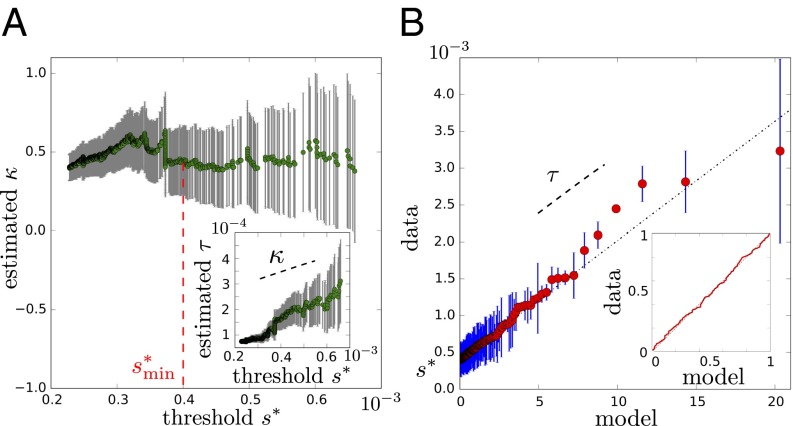
Variation and selection are the core principles of Darwinian evolution, but quantitatively relating the diversity of a population to its capacity to respond to selection is challenging. Here, we examine this problem at a molecular level in the context of populations of partially randomized proteins selected for binding to well-defined targets. We built several minimal protein libraries, screened them in vitro by phage display, and analyzed their response to selection by high-throughput sequencing. A statistical analysis of the results reveals two main findings. First, libraries with the same sequence diversity but built around different "frameworks" typically have vastly different responses; second, the distribution of responses of the best binders in a library follows a simple scaling law. We show how an elementary probabilistic model based on extreme value theory rationalizes the latter finding. Our results have implications for designing synthetic protein libraries, estimating the density of functional biomolecules in sequence space, characterizing diversity in natural populations, and experimentally investigating evolvability (i.e., the potential for future evolution).
Keywords: antibodies; biological diversity; directed evolution; extreme values; phage display.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Magurran AE. Measuring Biological Diversity. Wiley; New York: 2013. - PubMed
-
- Zhao H, Arnold FH. Combinatorial protein design: Strategies for screening protein libraries. Curr Opin Struct Biol. 1997;7(4):480–485. - PubMed
-
- Wong TS, Zhurina D, Schwaneberg U. The diversity challenge in directed protein evolution. Comb Chem High Throughput Screen. 2006;9(4):271–288. - PubMed
-
- Padlan EA. Anatomy of the antibody molecule. Mol Immunol. 1994;31(3):169–217. - PubMed
-
- Urvoas A, Valerio-Lepiniec M, Minard P. Artificial proteins from combinatorial approaches. Trends Biotechnol. 2012;30(10):512–520. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
