

Spectrum and Prevalence of CALM1-, CALM2-, and CALM3-Encoded Calmodulin Variants in Long QT Syndrome and Functional Characterization of a Novel Long QT Syndrome-Associated Calmodulin Missense Variant, E141G
- PMID: 26969752
- PMCID: PMC4907364
- DOI: 10.1161/CIRCGENETICS.115.001323
Spectrum and Prevalence of CALM1-, CALM2-, and CALM3-Encoded Calmodulin Variants in Long QT Syndrome and Functional Characterization of a Novel Long QT Syndrome-Associated Calmodulin Missense Variant, E141G
Abstract
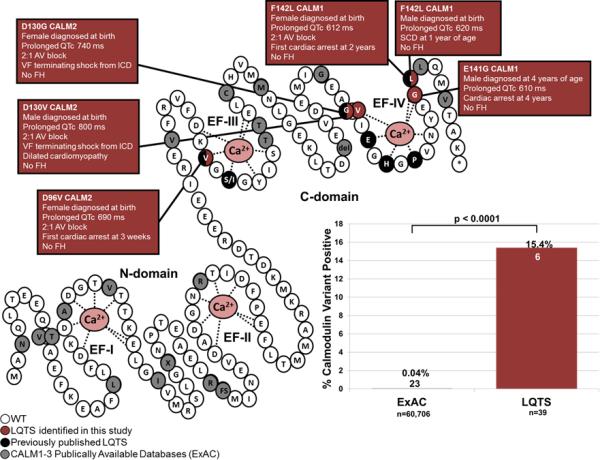
Background: Calmodulin (CaM) is encoded by 3 genes, CALM1, CALM2, and CALM3, all of which harbor pathogenic variants linked to long QT syndrome (LQTS) with early and severe expressivity. These LQTS-causative variants reduce CaM affinity to Ca(2+) and alter the properties of the cardiac L-type calcium channel (CaV1.2). CaM also modulates NaV1.5 and the ryanodine receptor, RyR2. All these interactions may play a role in disease pathogenesis. Here, we determine the spectrum and prevalence of pathogenic CaM variants in a cohort of genetically elusive LQTS, and functionally characterize the novel variants.
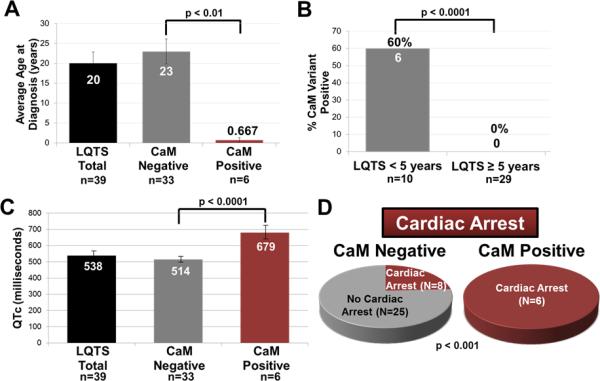
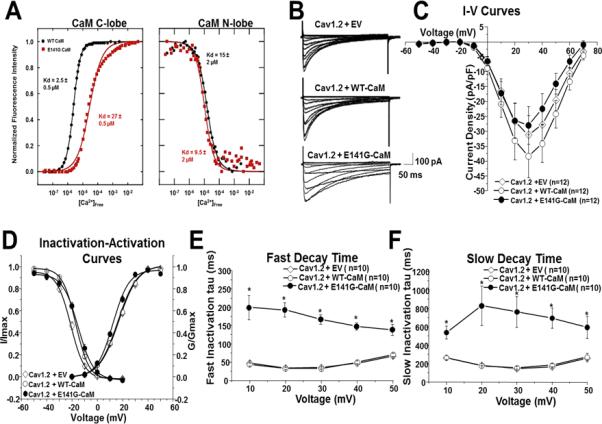
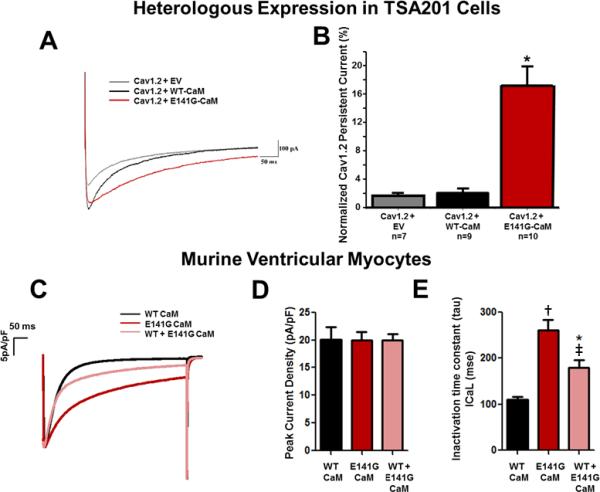
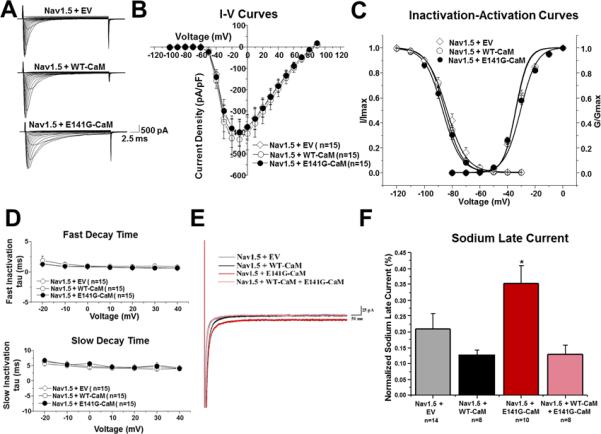
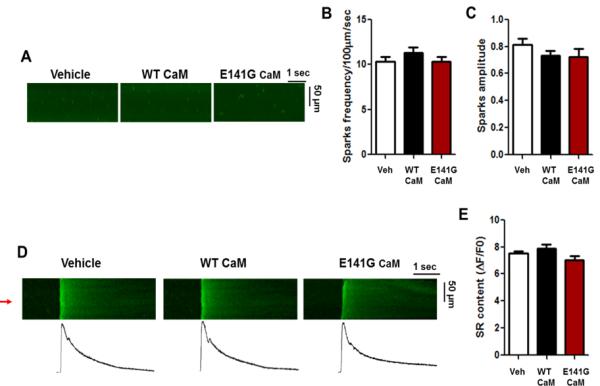
Methods and results: Thirty-eight genetically elusive LQTS cases underwent whole-exome sequencing to identify CaM variants. Nonsynonymous CaM variants were over-represented significantly in this heretofore LQTS cohort (13.2%) compared with exome aggregation consortium (0.04%; P<0.0001). When the clinical sequelae of these 5 CaM-positive cases were compared with the 33 CaM-negative cases, CaM-positive cases had a more severe phenotype with an average age of onset of 10 months, an average corrected QT interval of 676 ms, and a high prevalence of cardiac arrest. Functional characterization of 1 novel variant, E141G-CaM, revealed an 11-fold reduction in Ca(2+)-binding affinity and a functionally dominant loss of inactivation in CaV1.2, mild accentuation in NaV1.5 late current, but no effect on intracellular RyR2-mediated calcium release.
Conclusions: Overall, 13% of our genetically elusive LQTS cohort harbored nonsynonymous variants in CaM. Genetic testing of CALM1-3 should be pursued for individuals with LQTS, especially those with early childhood cardiac arrest, extreme QT prolongation, and a negative family history.
Keywords: L-type calcium channels; calmodulin; long QT syndrome; ryanodine receptor; sodium channels.
© 2016 American Heart Association, Inc.
Figures
References
-
- Levitan IB. It is calmodulin after all! Mediator of the calcium modulation of multiple ion channels. Neuron. 1999;22:645–648. - PubMed
-
- Berchtold M, Egli R, Rhyner J, Hameister H, Strehler E. Localization of the human bona fide calmodulin genes calm1, calm2, and calm3 to chromosomes 14q24-q31, 2p21.1-p21.3, and 19q13.2-q13.3. Genomics. 1993;16:461–465. - PubMed
-
- Fischer R, Koller M, Flura M, Mathews S, Strehler-Page M, Krebs J, et al. Multiple divergent mrnas code for a single human calmodulin. J Biol Chem. 1998;263:17055–17062. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
