

Membrane remodeling and mechanics: Experiments and simulations of α-Synuclein
- PMID: 26972046
- PMCID: PMC5081225
- DOI: 10.1016/j.bbamem.2016.03.012
Membrane remodeling and mechanics: Experiments and simulations of α-Synuclein
Abstract
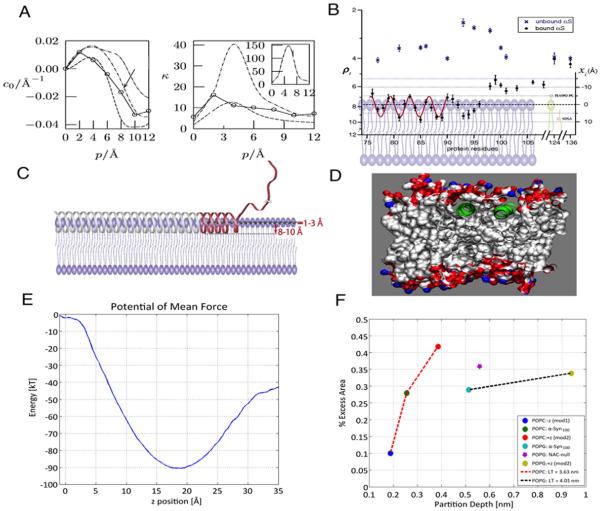
We review experimental and simulation approaches that have been used to determine curvature generation and remodeling of lipid bilayers by membrane-bending proteins. Particular emphasis is placed on the complementary approaches used to study α-Synuclein (αSyn), a major protein involved in Parkinson's disease (PD). Recent cellular and biophysical experiments have shown that the protein 1) deforms the native structure of mitochondrial and model membranes; and 2) inhibits vesicular fusion. Today's advanced experimental and computational technology has made it possible to quantify these protein-induced changes in membrane shape and material properties. Collectively, experiments, theory and multi-scale simulation techniques have established the key physical determinants of membrane remodeling and rigidity: protein binding energy, protein partition depth, protein density, and membrane tension. Despite the exciting and significant progress made in recent years in these areas, challenges remain in connecting biophysical insights to the cellular processes that lead to disease. This article is part of a Special Issue entitled: Membrane Proteins edited by J.C. Gumbart and Sergei Noskov.
Keywords: Bilayer rigidity; Coarse-grained molecular dynamics (CGMD); Membrane curvature; Parkinson's disease; Synaptic vesicles; Tubulation; α-Synuclein.
Copyright © 2016. Published by Elsevier B.V.
Figures







References
-
- Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng Y, Finkbeiner S, Nussbaum RL, Masliah E, Edwards RH. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011;286:20710–20726. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
