

GenSeed-HMM: A Tool for Progressive Assembly Using Profile HMMs as Seeds and its Application in Alpavirinae Viral Discovery from Metagenomic Data
- PMID: 26973638
- PMCID: PMC4777721
- DOI: 10.3389/fmicb.2016.00269
GenSeed-HMM: A Tool for Progressive Assembly Using Profile HMMs as Seeds and its Application in Alpavirinae Viral Discovery from Metagenomic Data
Abstract
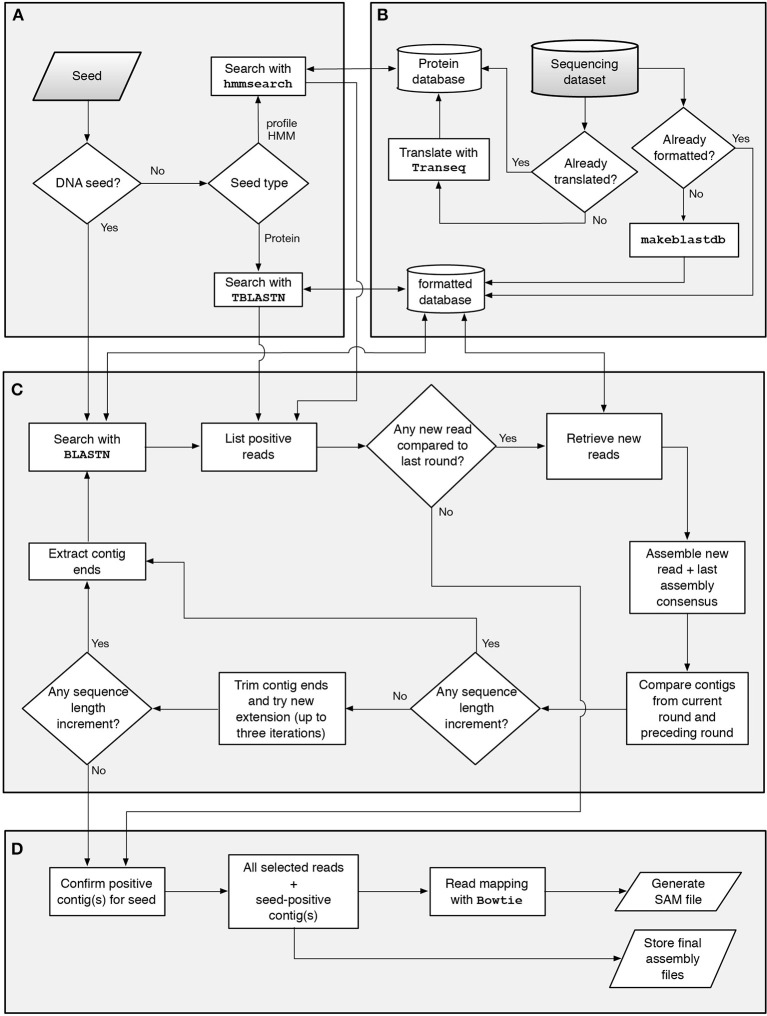
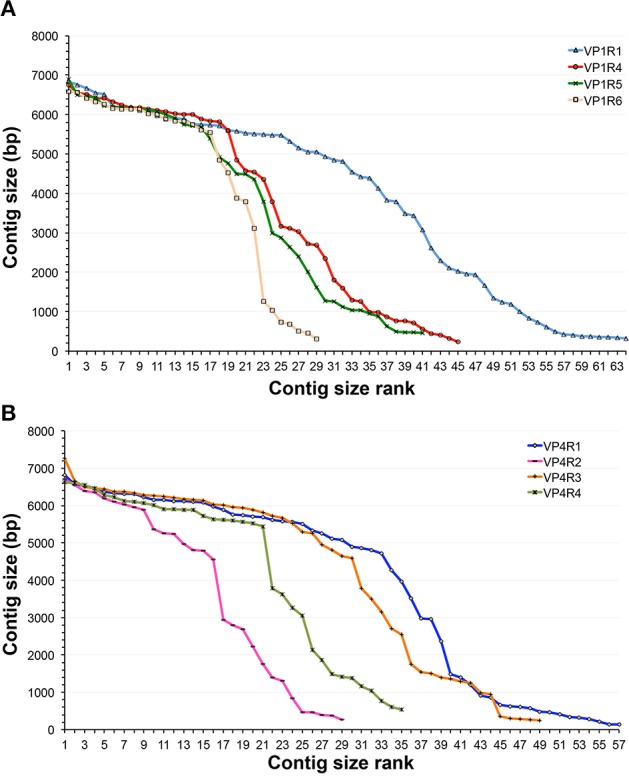
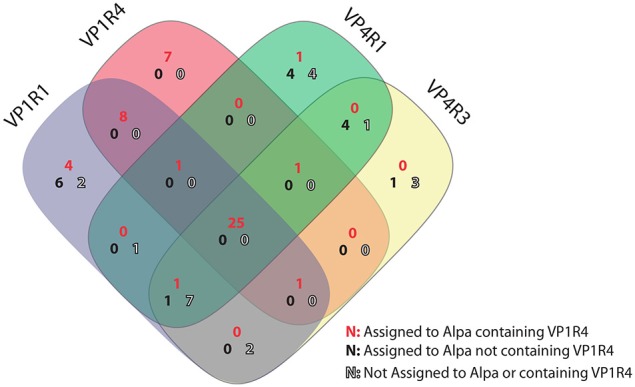
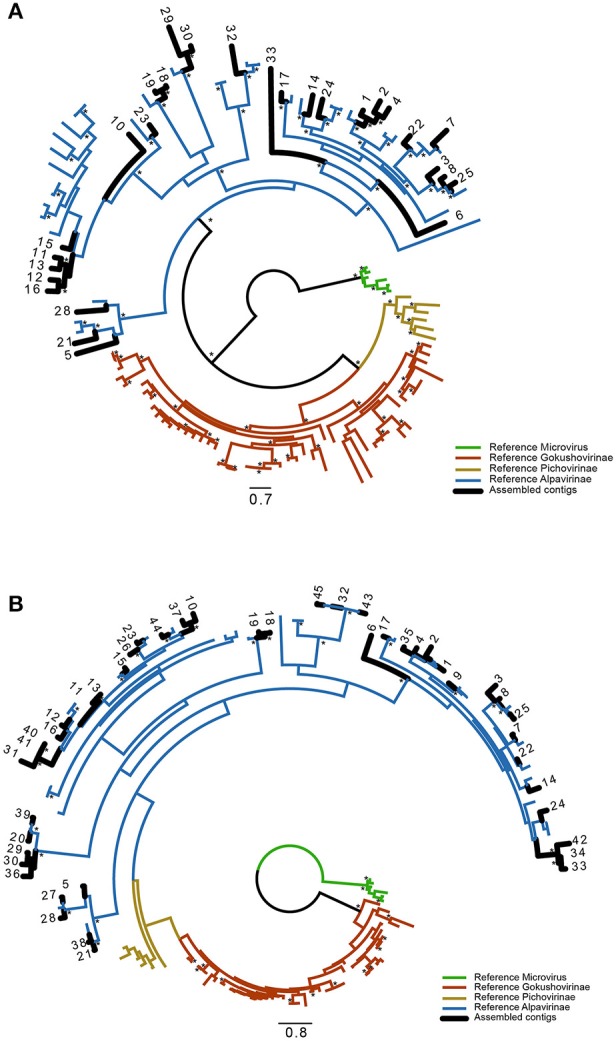
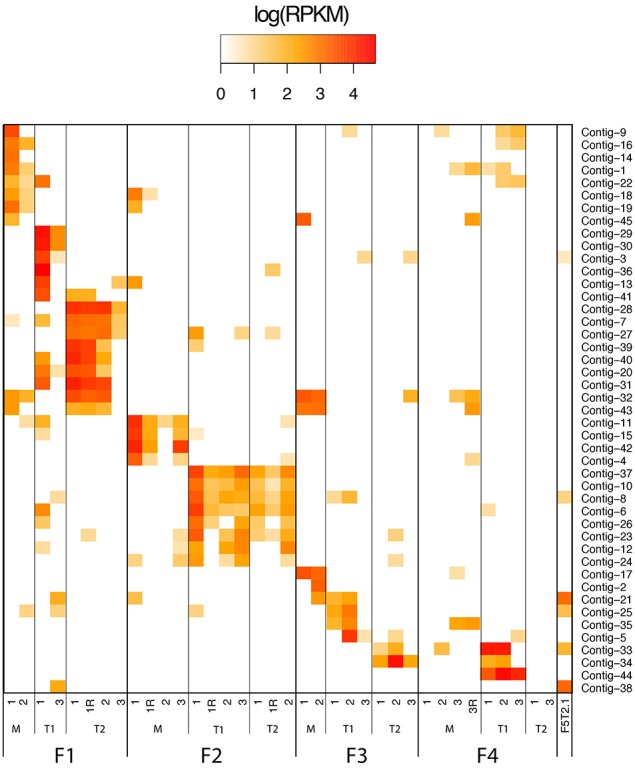
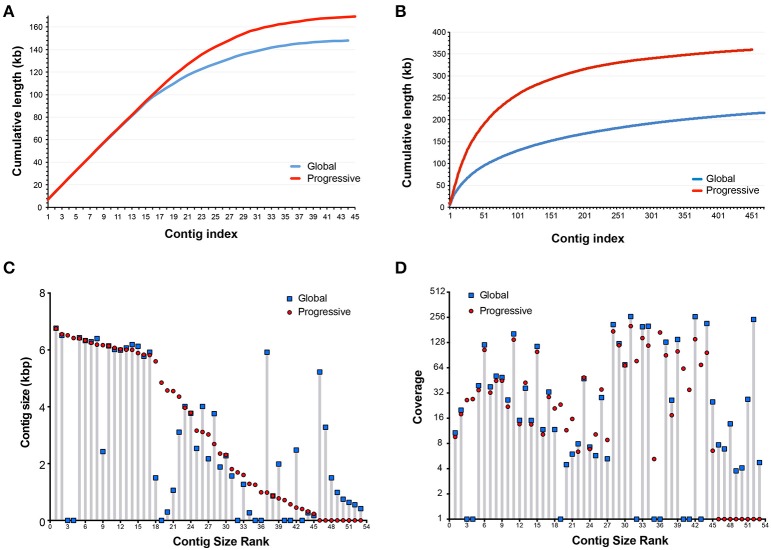
This work reports the development of GenSeed-HMM, a program that implements seed-driven progressive assembly, an approach to reconstruct specific sequences from unassembled data, starting from short nucleotide or protein seed sequences or profile Hidden Markov Models (HMM). The program can use any one of a number of sequence assemblers. Assembly is performed in multiple steps and relatively few reads are used in each cycle, consequently the program demands low computational resources. As a proof-of-concept and to demonstrate the power of HMM-driven progressive assemblies, GenSeed-HMM was applied to metagenomic datasets in the search for diverse ssDNA bacteriophages from the recently described Alpavirinae subfamily. Profile HMMs were built using Alpavirinae-specific regions from multiple sequence alignments (MSA) using either the viral protein 1 (VP1; major capsid protein) or VP4 (genome replication initiation protein). These profile HMMs were used by GenSeed-HMM (running Newbler assembler) as seeds to reconstruct viral genomes from sequencing datasets of human fecal samples. All contigs obtained were annotated and taxonomically classified using similarity searches and phylogenetic analyses. The most specific profile HMM seed enabled the reconstruction of 45 partial or complete Alpavirinae genomic sequences. A comparison with conventional (global) assembly of the same original dataset, using Newbler in a standalone execution, revealed that GenSeed-HMM outperformed global genomic assembly in several metrics employed. This approach is capable of detecting organisms that have not been used in the construction of the profile HMM, which opens up the possibility of diagnosing novel viruses, without previous specific information, constituting a de novo diagnosis. Additional applications include, but are not limited to, the specific assembly of extrachromosomal elements such as plastid and mitochondrial genomes from metagenomic data. Profile HMM seeds can also be used to reconstruct specific protein coding genes for gene diversity studies, and to determine all possible gene variants present in a metagenomic sample. Such surveys could be useful to detect the emergence of drug-resistance variants in sensitive environments such as hospitals and animal production facilities, where antibiotics are regularly used. Finally, GenSeed-HMM can be used as an adjunct for gap closure on assembly finishing projects, by using multiple contig ends as anchored seeds.
Keywords: Alpavirinae; de novo diagnosis; metagenomic analysis; sequence assembly; viral discovery.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous