

Multilocus Phylogeography and Species Delimitation in the Cumberland Plateau Salamander, Plethodon kentucki: Incongruence among Data Sets and Methods
- PMID: 26974148
- PMCID: PMC4790894
- DOI: 10.1371/journal.pone.0150022
Multilocus Phylogeography and Species Delimitation in the Cumberland Plateau Salamander, Plethodon kentucki: Incongruence among Data Sets and Methods
Abstract
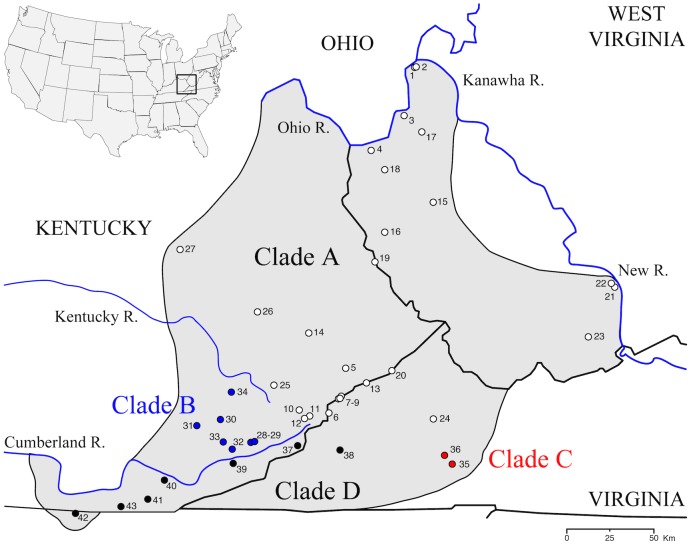
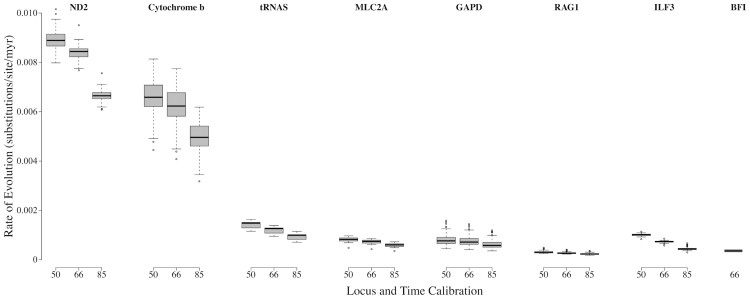
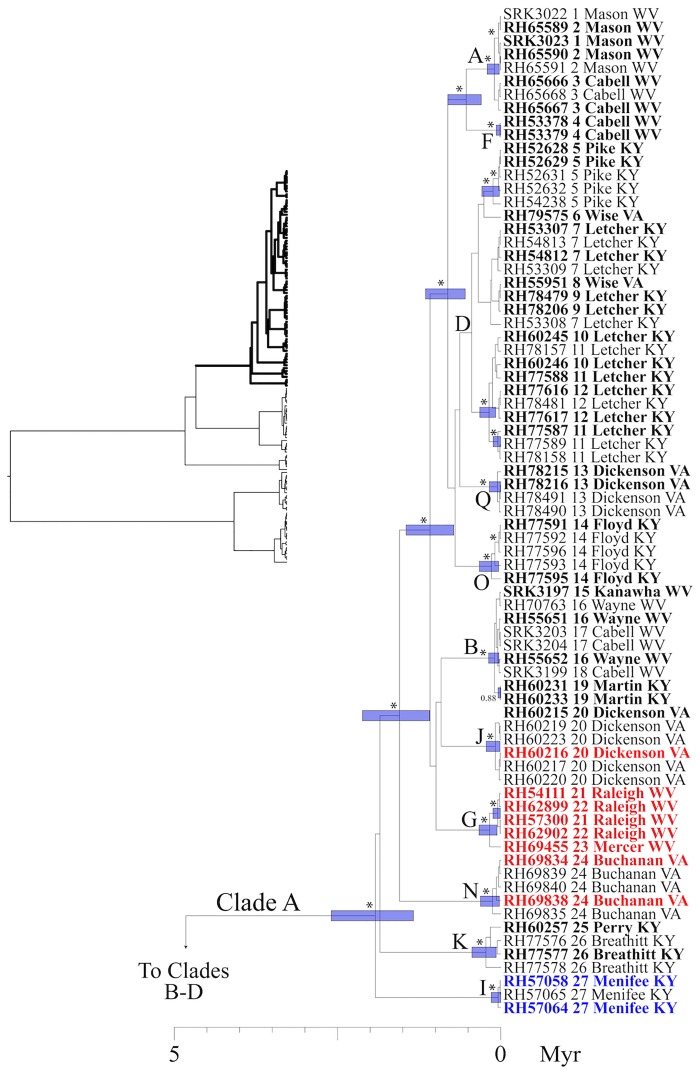
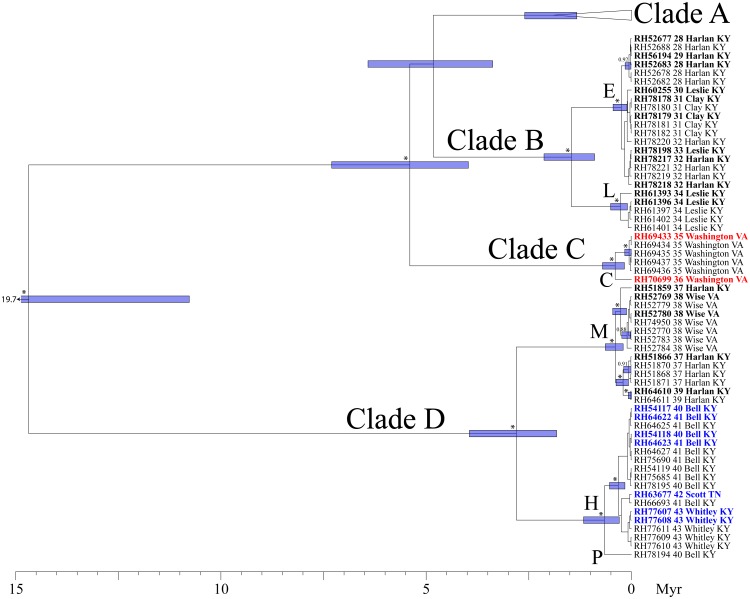
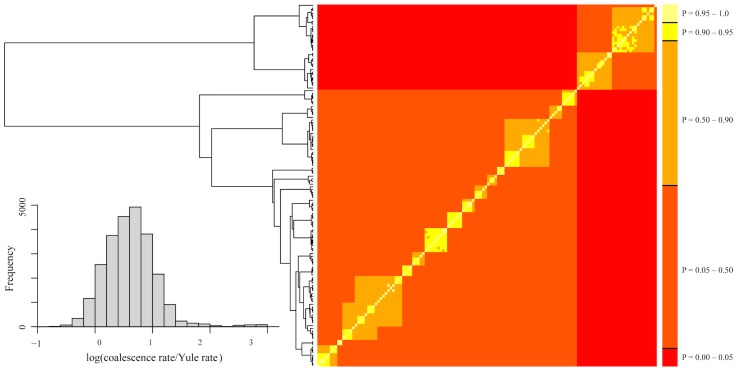
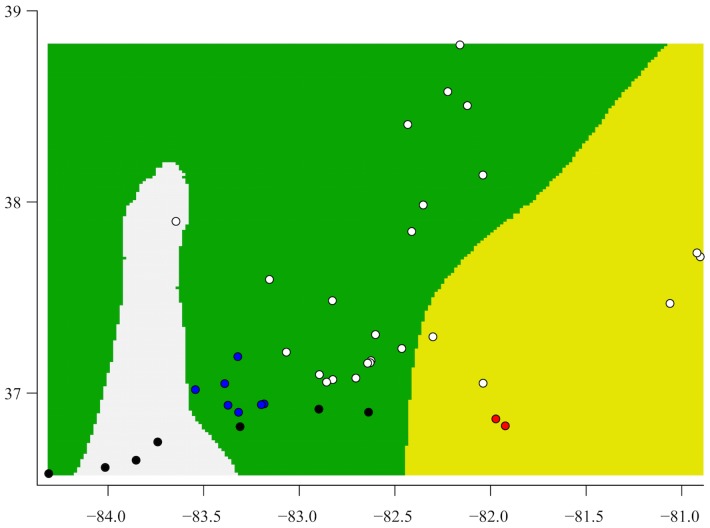
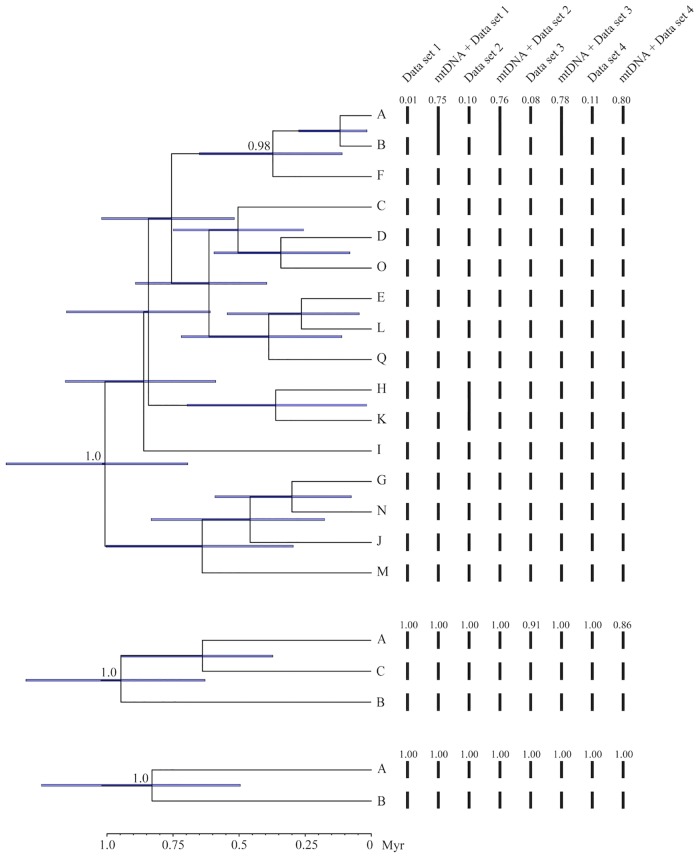
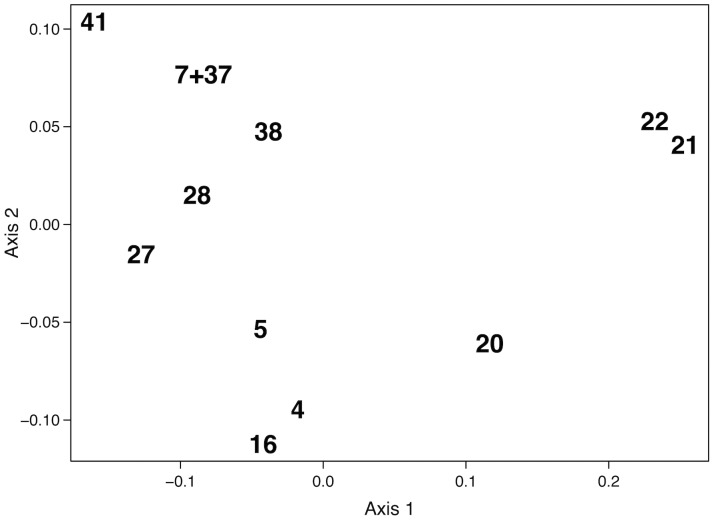
Species are a fundamental unit of biodiversity, yet can be challenging to delimit objectively. This is particularly true of species complexes characterized by high levels of population genetic structure, hybridization between genetic groups, isolation by distance, and limited phenotypic variation. Previous work on the Cumberland Plateau Salamander, Plethodon kentucki, suggested that it might constitute a species complex despite occupying a relatively small geographic range. To examine this hypothesis, we sampled 135 individuals from 43 populations, and used four mitochondrial loci and five nuclear loci (5693 base pairs) to quantify phylogeographic structure and probe for cryptic species diversity. Rates of evolution for each locus were inferred using the multidistribute package, and time calibrated gene trees and species trees were inferred using BEAST 2 and *BEAST 2, respectively. Because the parameter space relevant for species delimitation is large and complex, and all methods make simplifying assumptions that may lead them to fail, we conducted an array of analyses. Our assumption was that strongly supported species would be congruent across methods. Putative species were first delimited using a Bayesian implementation of the GMYC model (bGMYC), Geneland, and Brownie. We then validated these species using the genealogical sorting index and BPP. We found substantial phylogeographic diversity using mtDNA, including four divergent clades and an inferred common ancestor at 14.9 myr (95% HPD: 10.8-19.7 myr). By contrast, this diversity was not corroborated by nuclear sequence data, which exhibited low levels of variation and weak phylogeographic structure. Species trees estimated a far younger root than did the mtDNA data, closer to 1.0 myr old. Mutually exclusive putative species were identified by the different approaches. Possible causes of data set discordance, and the problem of species delimitation in complexes with high levels of population structure and introgressive hybridization, are discussed.
Conflict of interest statement
Figures
References
-
- Sites JW, Marshall JC. Delimiting species: a renaissance issue in systematic biology. Trends Ecol Evol. 2003; 18: 462–470. 10.1016/S0169-5347(03)00184-8 - DOI
-
- Sites JW, Marshall JC. Operational criteria for delimiting species. Ann Rev Ecol Evol Syst. 2004; 35: 199–227. 10.1146/annurev.ecolsys.35.112202.130128 - DOI
-
- Kuchta S, Wake DB. Wherefore and wither the ring species? Copeia 2016; In press.
-
- Camargo A, Sites JW. Species delimitation: a decade after the renaissance In: Pavlinov IY, editor. The Species Problem: Ongoing Issues. InTech—Open Access publisher, Rijeka, Croatia; 2013. 10.5772/52664 - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
