

Axial Spondylometaphyseal Dysplasia Is Caused by C21orf2 Mutations
- PMID: 26974433
- PMCID: PMC4790905
- DOI: 10.1371/journal.pone.0150555
Axial Spondylometaphyseal Dysplasia Is Caused by C21orf2 Mutations
Abstract
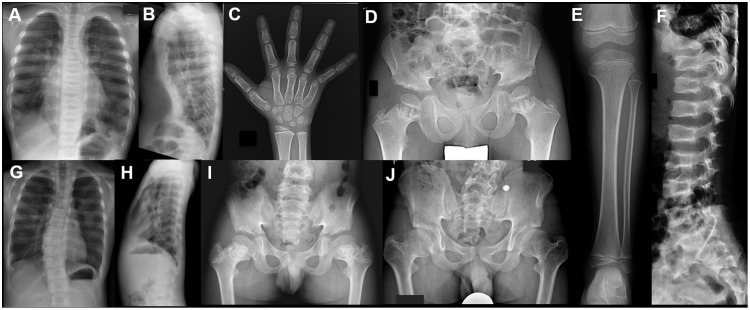
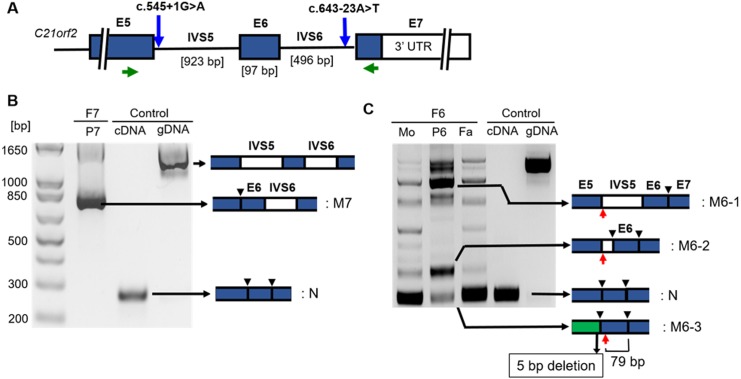
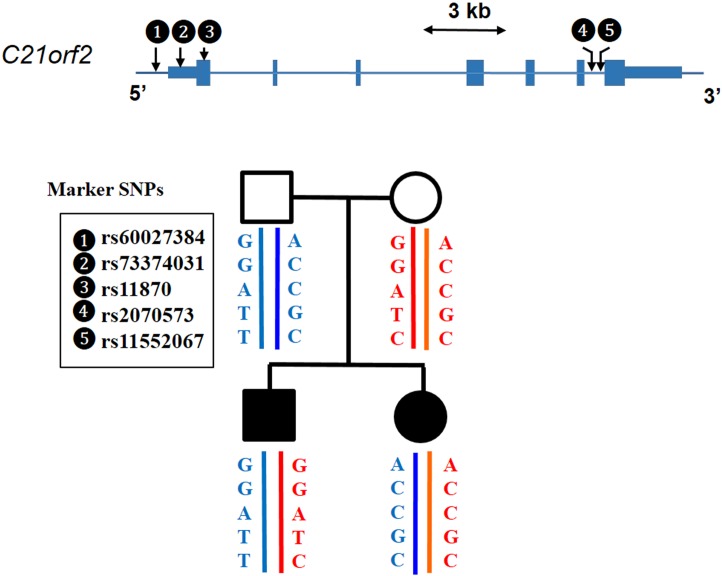
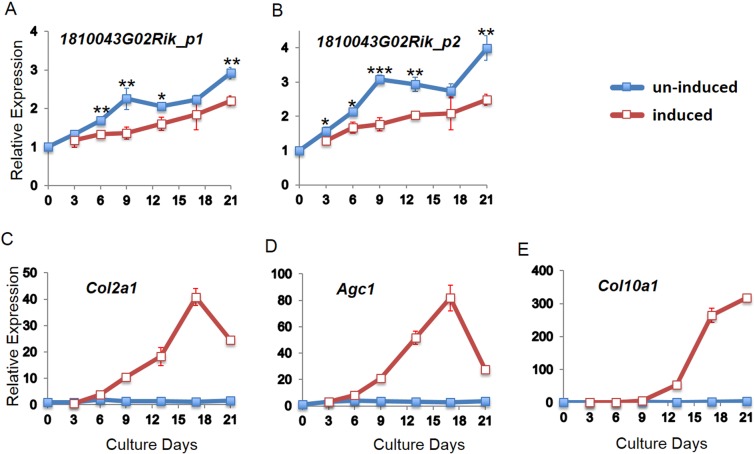
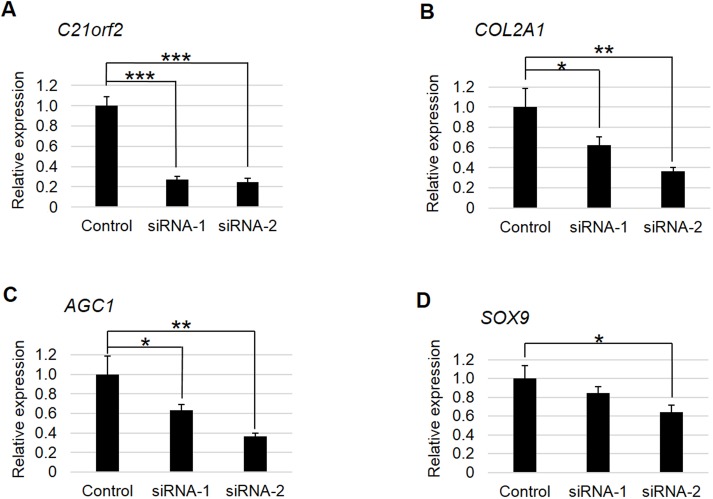
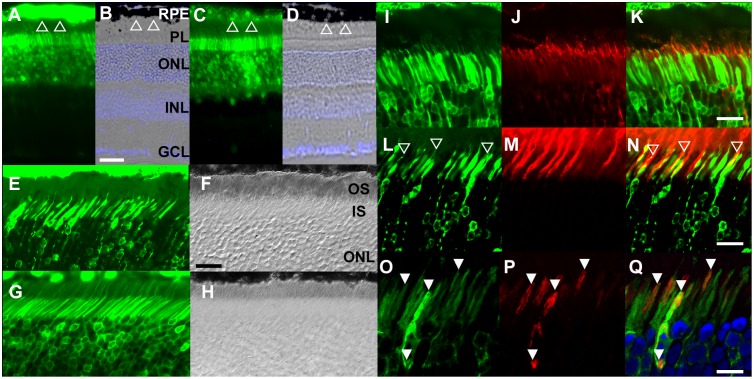
Axial spondylometaphyseal dysplasia (axial SMD) is an autosomal recessive disease characterized by dysplasia of axial skeleton and retinal dystrophy. We conducted whole exome sequencing and identified C21orf2 (chromosome 21 open reading frame 2) as a disease gene for axial SMD. C21orf2 mutations have been recently found to cause isolated retinal degeneration and Jeune syndrome. We found a total of five biallelic C21orf2 mutations in six families out of nine: three missense and two splicing mutations in patients with various ethnic backgrounds. The pathogenic effects of the splicing (splice-site and branch-point) mutations were confirmed on RNA level, which showed complex patterns of abnormal splicing. C21orf2 mutations presented with a wide range of skeletal phenotypes, including cupped and flared anterior ends of ribs, lacy ilia and metaphyseal dysplasia of proximal femora. Analysis of patients without C21orf2 mutation indicated genetic heterogeneity of axial SMD. Functional data in chondrocyte suggest C21orf2 is implicated in cartilage differentiation. C21orf2 protein was localized to the connecting cilium of the cone and rod photoreceptors, confirming its significance in retinal function. Our study indicates that axial SMD is a member of a unique group of ciliopathy affecting skeleton and retina.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
