

A cationic tetrapyrrole inhibits toxic activities of the cellular prion protein
- PMID: 26976106
- PMCID: PMC4791597
- DOI: 10.1038/srep23180
A cationic tetrapyrrole inhibits toxic activities of the cellular prion protein
Abstract
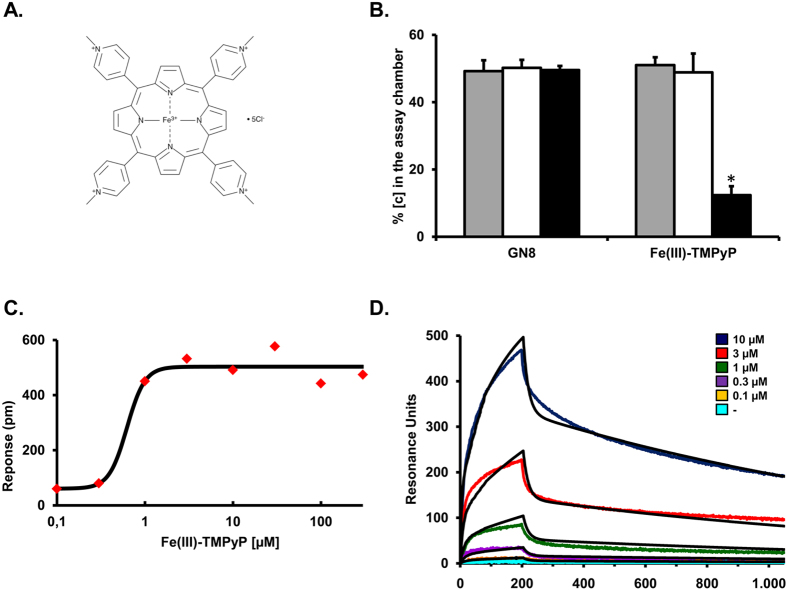
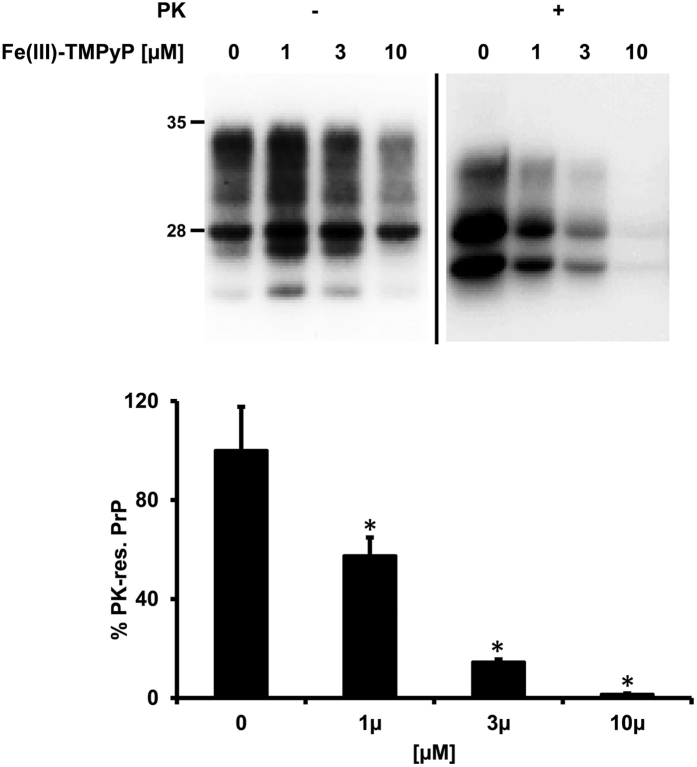
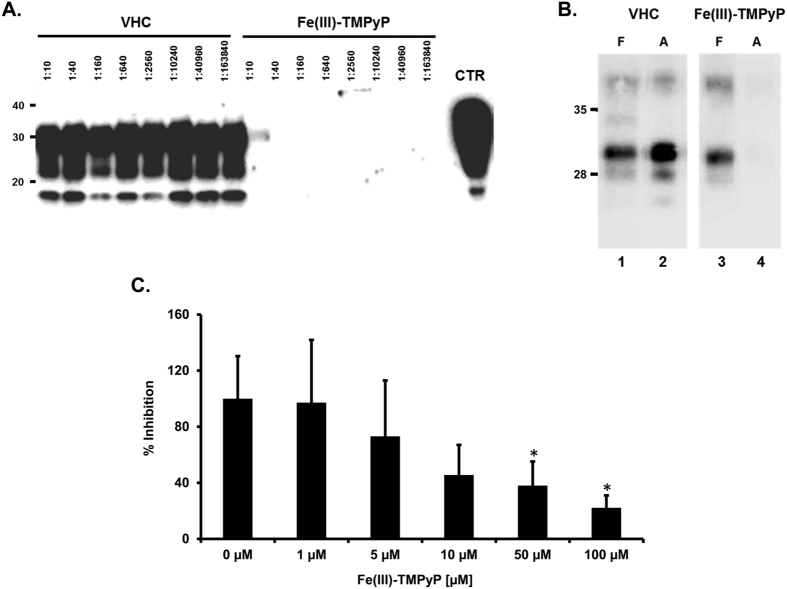
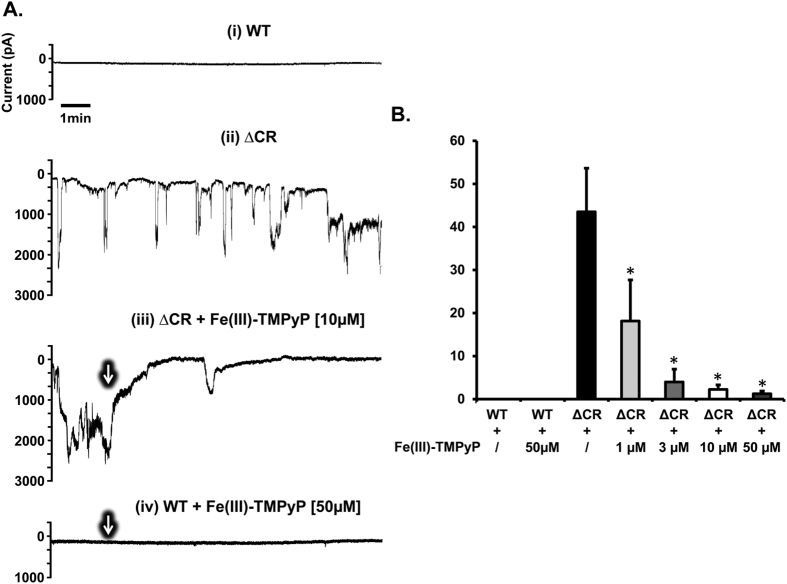
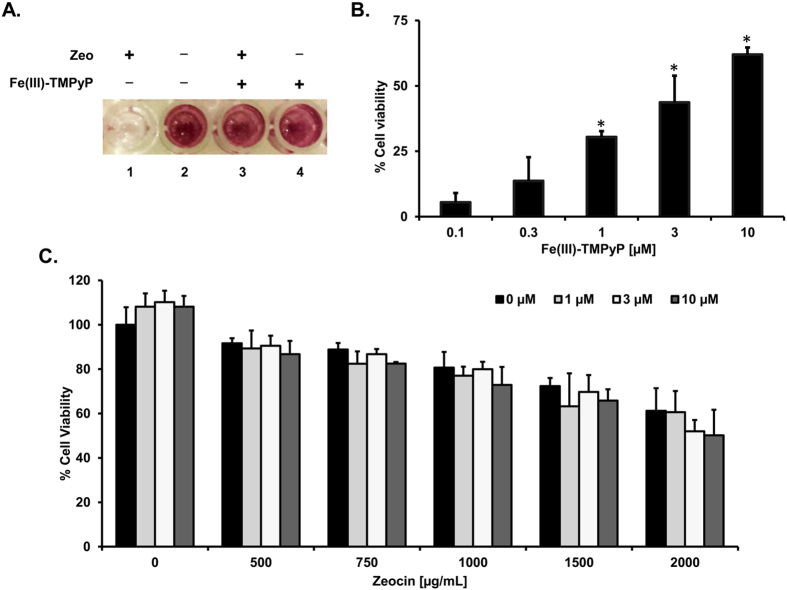
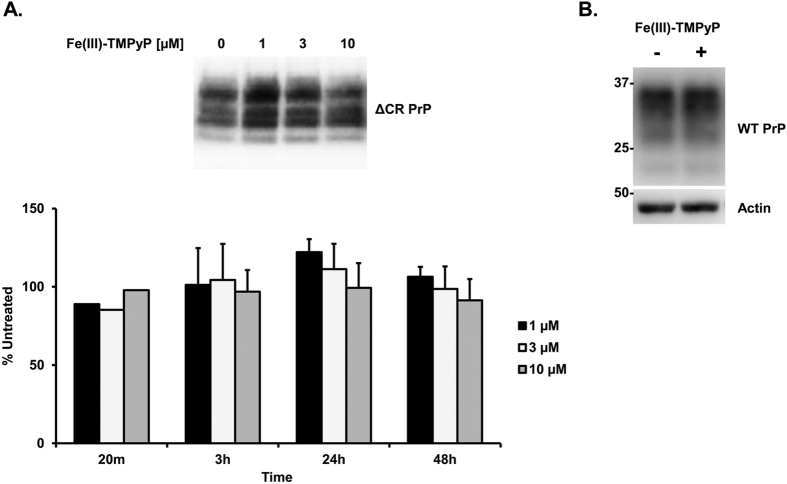
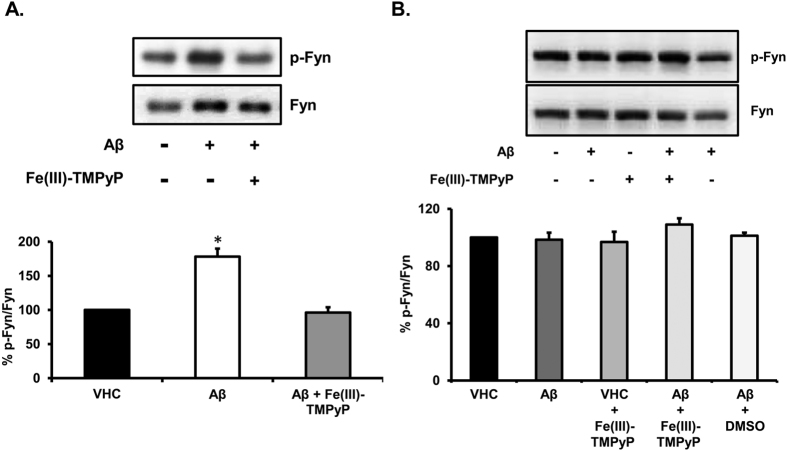
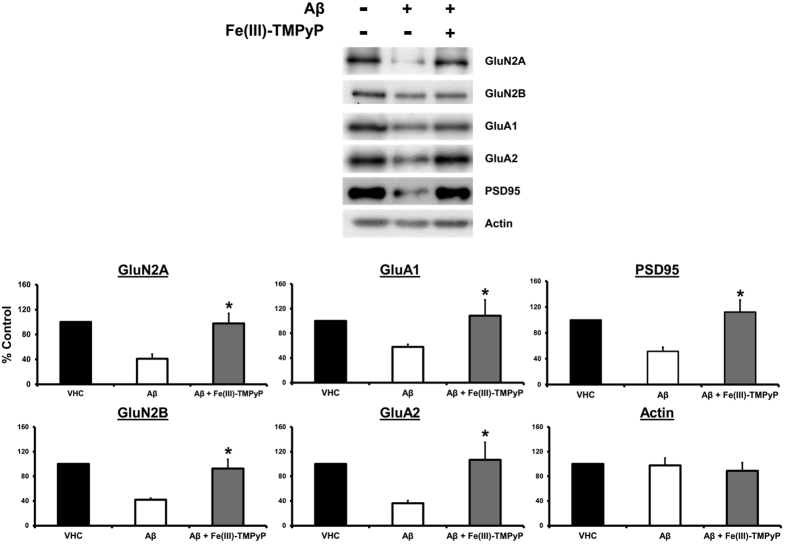
Prion diseases are rare neurodegenerative conditions associated with the conformational conversion of the cellular prion protein (PrP(C)) into PrP(Sc), a self-replicating isoform (prion) that accumulates in the central nervous system of affected individuals. The structure of PrP(Sc) is poorly defined, and likely to be heterogeneous, as suggested by the existence of different prion strains. The latter represents a relevant problem for therapy in prion diseases, as some potent anti-prion compounds have shown strain-specificity. Designing therapeutics that target PrP(C) may provide an opportunity to overcome these problems. PrP(C) ligands may theoretically inhibit the replication of multiple prion strains, by acting on the common substrate of any prion replication reaction. Here, we characterized the properties of a cationic tetrapyrrole [Fe(III)-TMPyP], which was previously shown to bind PrP(C), and inhibit the replication of a mouse prion strain. We report that the compound is active against multiple prion strains in vitro and in cells. Interestingly, we also find that Fe(III)-TMPyP inhibits several PrP(C)-related toxic activities, including the channel-forming ability of a PrP mutant, and the PrP(C)-dependent synaptotoxicity of amyloid-β (Aβ) oligomers, which are associated with Alzheimer's Disease. These results demonstrate that molecules binding to PrP(C) may produce a dual effect of blocking prion replication and inhibiting PrP(C)-mediated toxicity.
Figures
Similar articles
-
High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer's disease.Brain. 2014 Mar;137(Pt 3):873-86. doi: 10.1093/brain/awt375. Epub 2014 Feb 10. Brain. 2014. PMID: 24519981
-
Interaction of Peptide Aptamers with Prion Protein Central Domain Promotes α-Cleavage of PrPC.Mol Neurobiol. 2018 Oct;55(10):7758-7774. doi: 10.1007/s12035-018-0944-9. Epub 2018 Feb 19. Mol Neurobiol. 2018. PMID: 29460268 Free PMC article.
-
Virus Infection, Genetic Mutations, and Prion Infection in Prion Protein Conversion.Int J Mol Sci. 2021 Nov 18;22(22):12439. doi: 10.3390/ijms222212439. Int J Mol Sci. 2021. PMID: 34830321 Free PMC article. Review.
-
Identification of a Compound That Disrupts Binding of Amyloid-β to the Prion Protein Using a Novel Fluorescence-based Assay.J Biol Chem. 2015 Jul 3;290(27):17020-8. doi: 10.1074/jbc.M115.637124. Epub 2015 May 20. J Biol Chem. 2015. PMID: 25995455 Free PMC article.
-
Heterogeneity and Architecture of Pathological Prion Protein Assemblies: Time to Revisit the Molecular Basis of the Prion Replication Process?Viruses. 2019 May 10;11(5):429. doi: 10.3390/v11050429. Viruses. 2019. PMID: 31083283 Free PMC article. Review.
Cited by
-
Unfolded and intermediate states of PrP play a key role in the mechanism of action of an antiprion chaperone.Proc Natl Acad Sci U S A. 2021 Mar 2;118(9):e2010213118. doi: 10.1073/pnas.2010213118. Proc Natl Acad Sci U S A. 2021. PMID: 33619087 Free PMC article.
-
Multimodal small-molecule screening for human prion protein binders.J Biol Chem. 2020 Sep 25;295(39):13516-13531. doi: 10.1074/jbc.RA120.014905. Epub 2020 Jul 28. J Biol Chem. 2020. PMID: 32723867 Free PMC article.
-
A Promising Antiprion Trimethoxychalcone Binds to the Globular Domain of the Cellular Prion Protein and Changes Its Cellular Location.Antimicrob Agents Chemother. 2018 Jan 25;62(2):e01441-17. doi: 10.1128/AAC.01441-17. Print 2018 Feb. Antimicrob Agents Chemother. 2018. PMID: 29133563 Free PMC article.
-
Pentosan polysulfate induces low-level persistent prion infection keeping measurable seeding activity without PrP-res detection in Fukuoka-1 infected cell cultures.Sci Rep. 2022 May 13;12(1):7923. doi: 10.1038/s41598-022-12049-z. Sci Rep. 2022. PMID: 35562591 Free PMC article.
-
The Compelling Demand for an Effective PrPC-Directed Therapy against Prion Diseases.ACS Med Chem Lett. 2020 Nov 2;11(11):2063-2067. doi: 10.1021/acsmedchemlett.0c00528. eCollection 2020 Nov 12. ACS Med Chem Lett. 2020. PMID: 33209189 Free PMC article.
References
-
- Collinge J. & Clarke A. R. A general model of prion strains and their pathogenicity. Science 318, 930–936 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
