

Aurone synthase is a catechol oxidase with hydroxylase activity and provides insights into the mechanism of plant polyphenol oxidases
- PMID: 26976571
- PMCID: PMC4822611
- DOI: 10.1073/pnas.1523575113
Aurone synthase is a catechol oxidase with hydroxylase activity and provides insights into the mechanism of plant polyphenol oxidases
Abstract
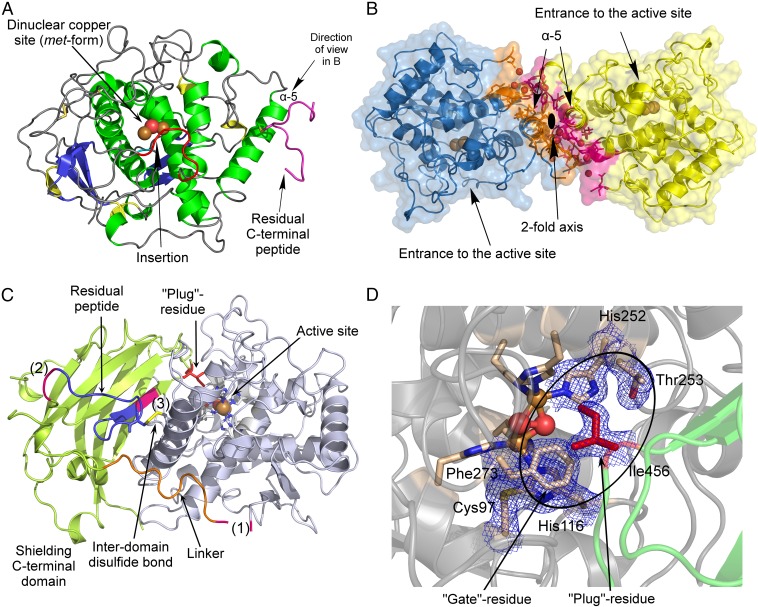
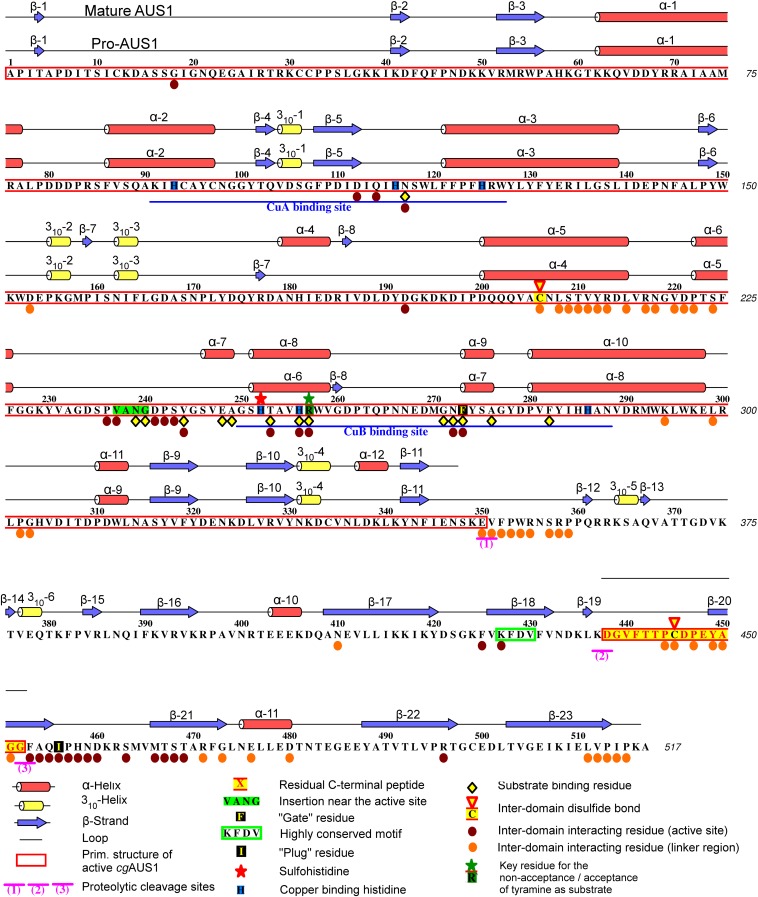
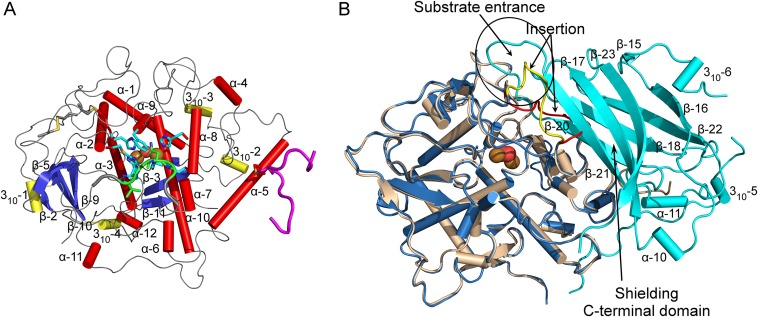
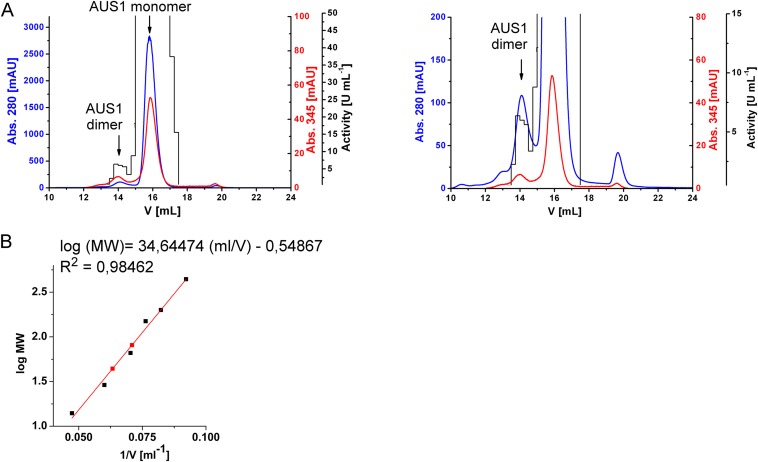
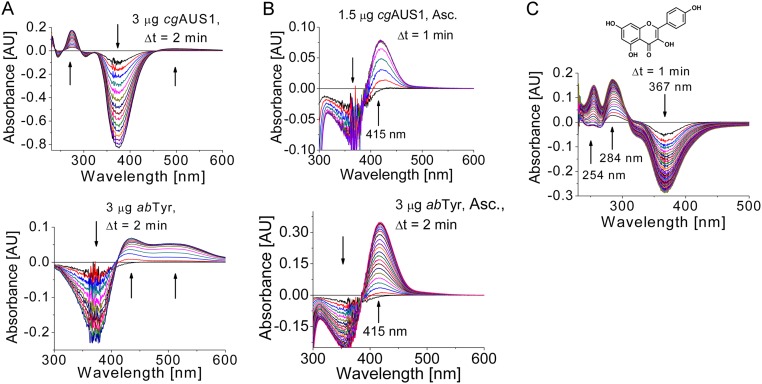
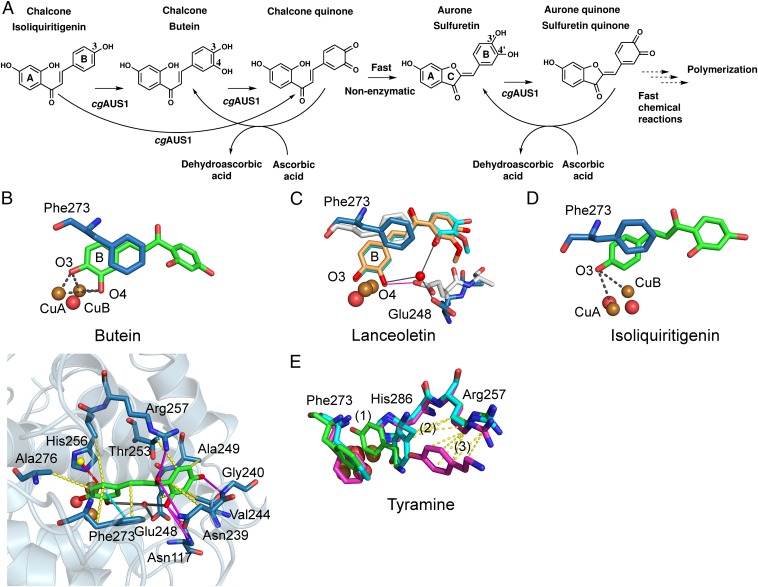
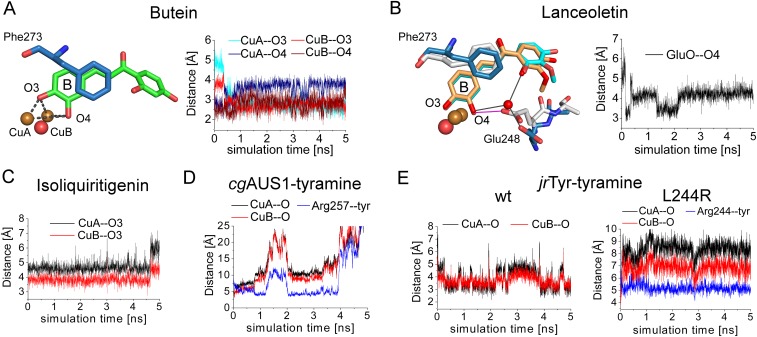
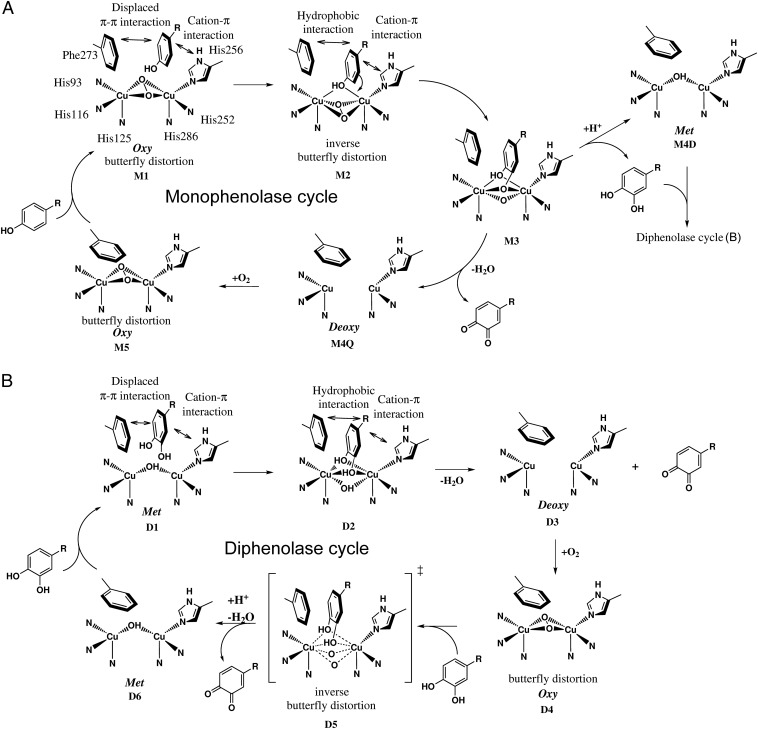
Tyrosinases and catechol oxidases belong to the family of polyphenol oxidases (PPOs). Tyrosinases catalyze theo-hydroxylation and oxidation of phenolic compounds, whereas catechol oxidases were so far defined to lack the hydroxylation activity and catalyze solely the oxidation of o-diphenolic compounds. Aurone synthase from Coreopsis grandiflora (AUS1) is a specialized plant PPO involved in the anabolic pathway of aurones. We present, to our knowledge, the first crystal structures of a latent plant PPO, its mature active and inactive form, caused by a sulfation of a copper binding histidine. Analysis of the latent proenzyme's interface between the shielding C-terminal domain and the main core provides insights into its activation mechanisms. As AUS1 did not accept common tyrosinase substrates (tyrosine and tyramine), the enzyme is classified as a catechol oxidase. However, AUS1 showed hydroxylase activity toward its natural substrate (isoliquiritigenin), revealing that the hydroxylase activity is not correlated with the acceptance of common tyrosinase substrates. Therefore, we propose that the hydroxylase reaction is a general functionality of PPOs. Molecular dynamics simulations of docked substrate-enzyme complexes were performed, and a key residue was identified that influences the plant PPO's acceptance or rejection of tyramine. Based on the evidenced hydroxylase activity and the interactions of specific residues with the substrates during the molecular dynamics simulations, a novel catalytic reaction mechanism for plant PPOs is proposed. The presented results strongly suggest that the physiological role of plant catechol oxidases were previously underestimated, as they might hydroxylate their--so far unknown--natural substrates in vivo.
Keywords: catechol oxidase; crystal structure; mechanism; type III copper protein; tyrosinase.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Mayer AM. Polyphenol oxidases in plants and fungi: Going places? A review. Phytochemistry. 2006;67(21):2318–2331. - PubMed
-
- King RS, Flurkey WH. Effects of limited proteolysis on broad bean polyphenoloxidase. J Sci Food Agric. 1987;41(3):231–240.
-
- Marusek CM, Trobaugh NM, Flurkey WH, Inlow JK. Comparative analysis of polyphenol oxidase from plant and fungal species. J Inorg Biochem. 2006;100(1):108–123. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
