

Characterization of Viral Communities of Biting Midges and Identification of Novel Thogotovirus Species and Rhabdovirus Genus
- PMID: 26978389
- PMCID: PMC4810267
- DOI: 10.3390/v8030077
Characterization of Viral Communities of Biting Midges and Identification of Novel Thogotovirus Species and Rhabdovirus Genus
Abstract
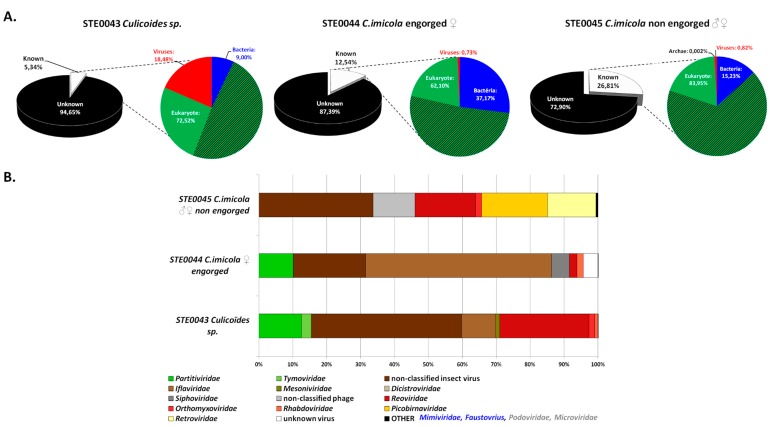
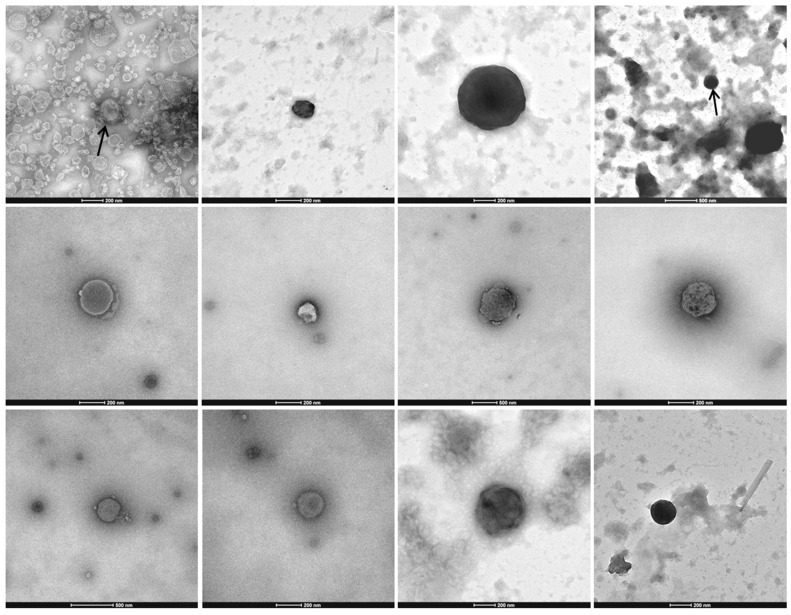
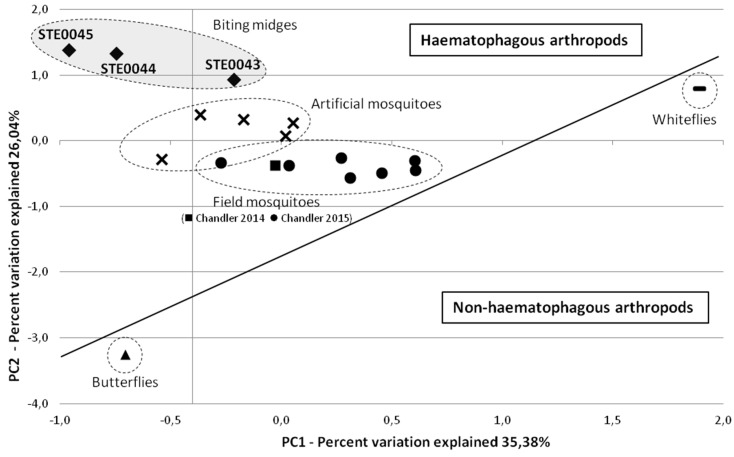
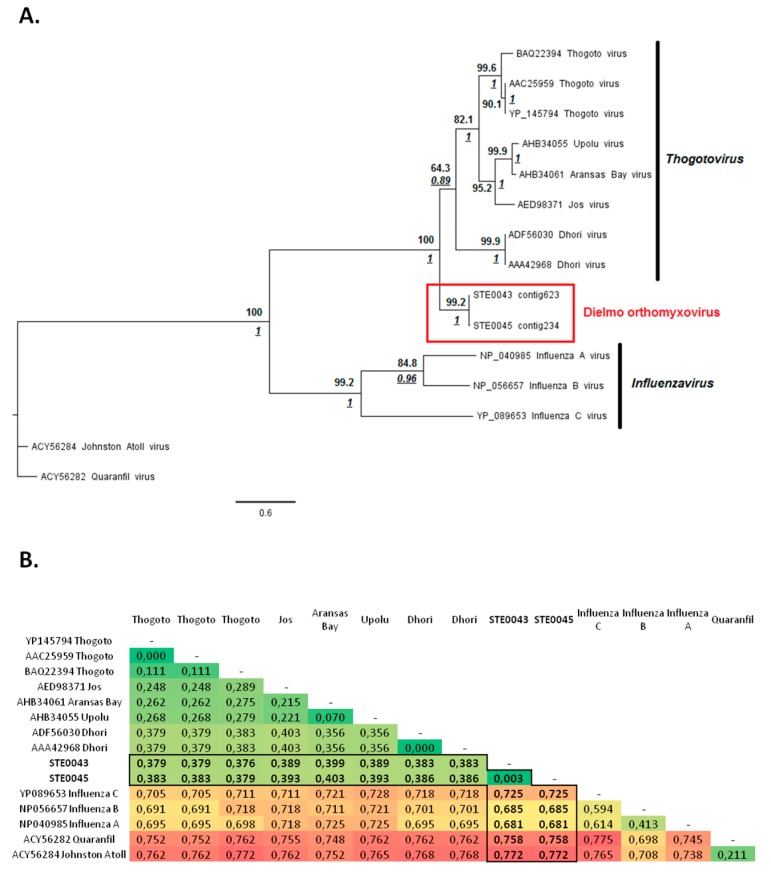
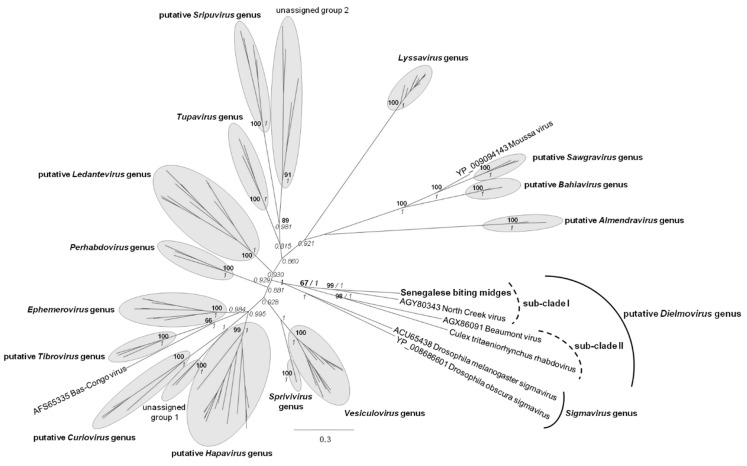
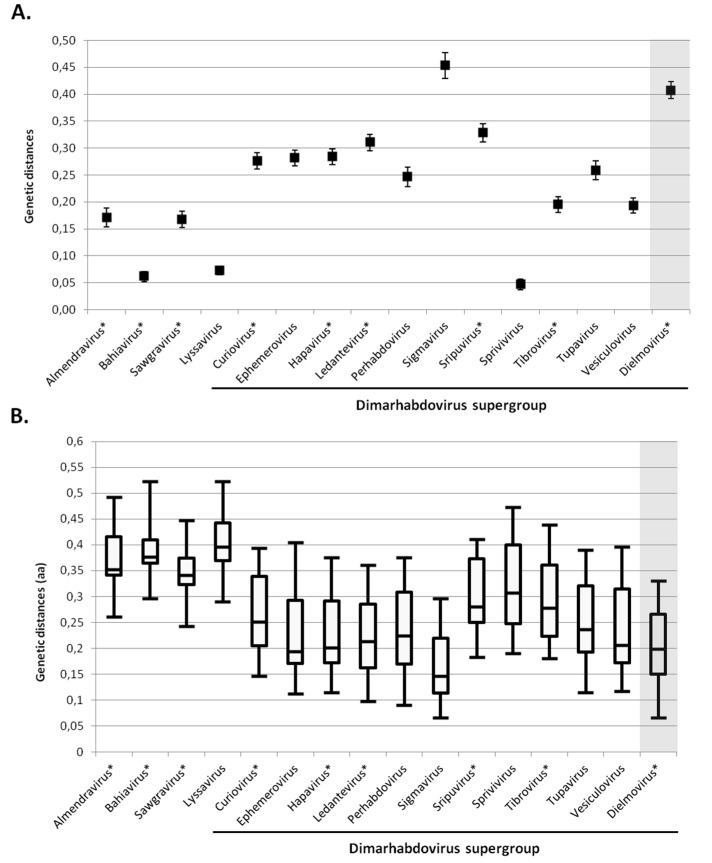
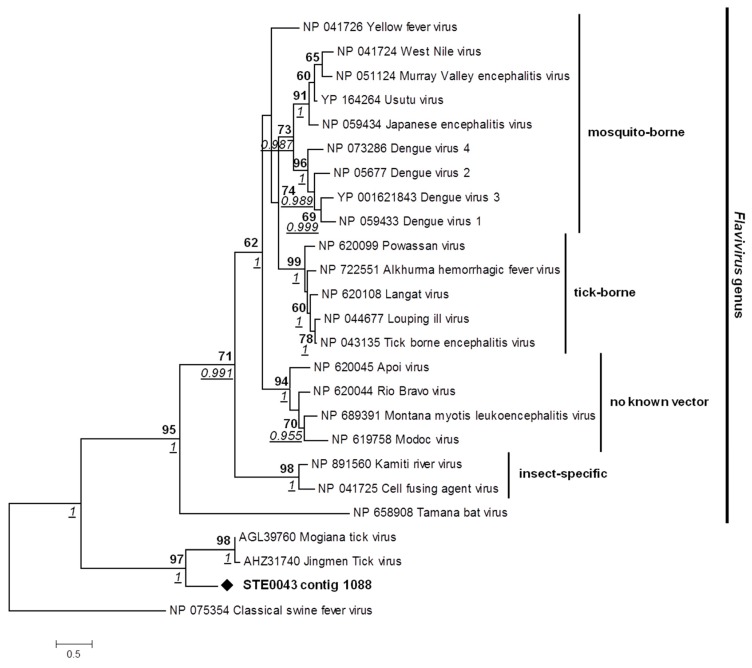
More than two thirds of emerging viruses are of zoonotic origin, and among them RNA viruses represent the majority. Ceratopogonidae (genus Culicoides) are well-known vectors of several viruses responsible for epizooties (bluetongue, epizootic haemorrhagic disease, etc.). They are also vectors of the only known virus infecting humans: the Oropouche virus. Female midges usually feed on a variety of hosts, leading to possible transmission of emerging viruses from animals to humans. In this context, we report here the analysis of RNA viral communities of Senegalese biting midges using next-generation sequencing techniques as a preliminary step toward the identification of potential viral biohazards. Sequencing of the RNA virome of three pools of Culicoides revealed the presence of a significant diversity of viruses infecting plants, insects and mammals. Several novel viruses were detected, including a novel Thogotovirus species, related but genetically distant from previously described tick-borne thogotoviruses. Novel rhabdoviruses were also detected, possibly constituting a novel Rhabdoviridae genus, and putatively restricted to insects. Sequences related to the major viruses transmitted by Culicoides, i.e., African horse sickness, bluetongue and epizootic haemorrhagic disease viruses were also detected. This study highlights the interest in monitoring the emergence and circulation of zoonoses and epizooties using their arthropod vectors.
Keywords: biting midges; epizooties; rhabdovirus; thogotovirus; viral metagenomics; zoonoses.
Figures
References
-
- Bishop-Lilly K.A., Turell M.J., Willner K.M., Butani A., Nolan N.M., Lentz S.M., Akmal A., Mateczun A., Brahmbhatt T.N., Sozhammannan S., et al. Arbovirus detection in insect vectors by rapid, high-throughput pyrosequencing. PLoS Negl. Trop. Dis. 2010;4 doi: 10.1371/journal.pntd.0000878. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
