

ncRNA orthologies in the vertebrate lineage
- PMID: 26980512
- PMCID: PMC4792531
- DOI: 10.1093/database/bav127
ncRNA orthologies in the vertebrate lineage
Abstract
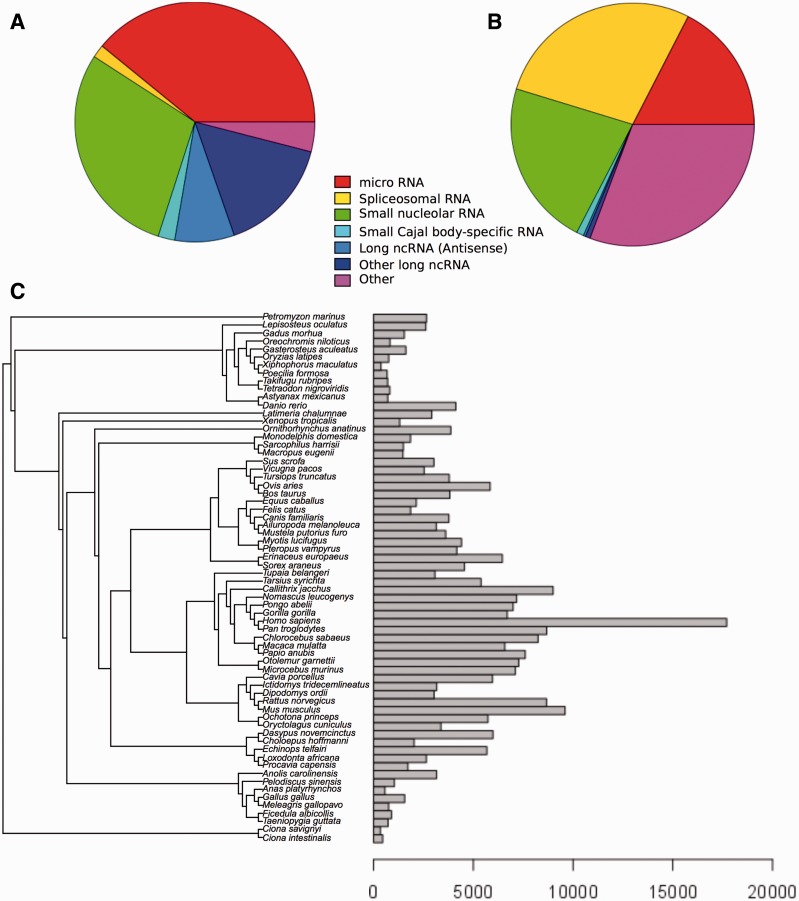
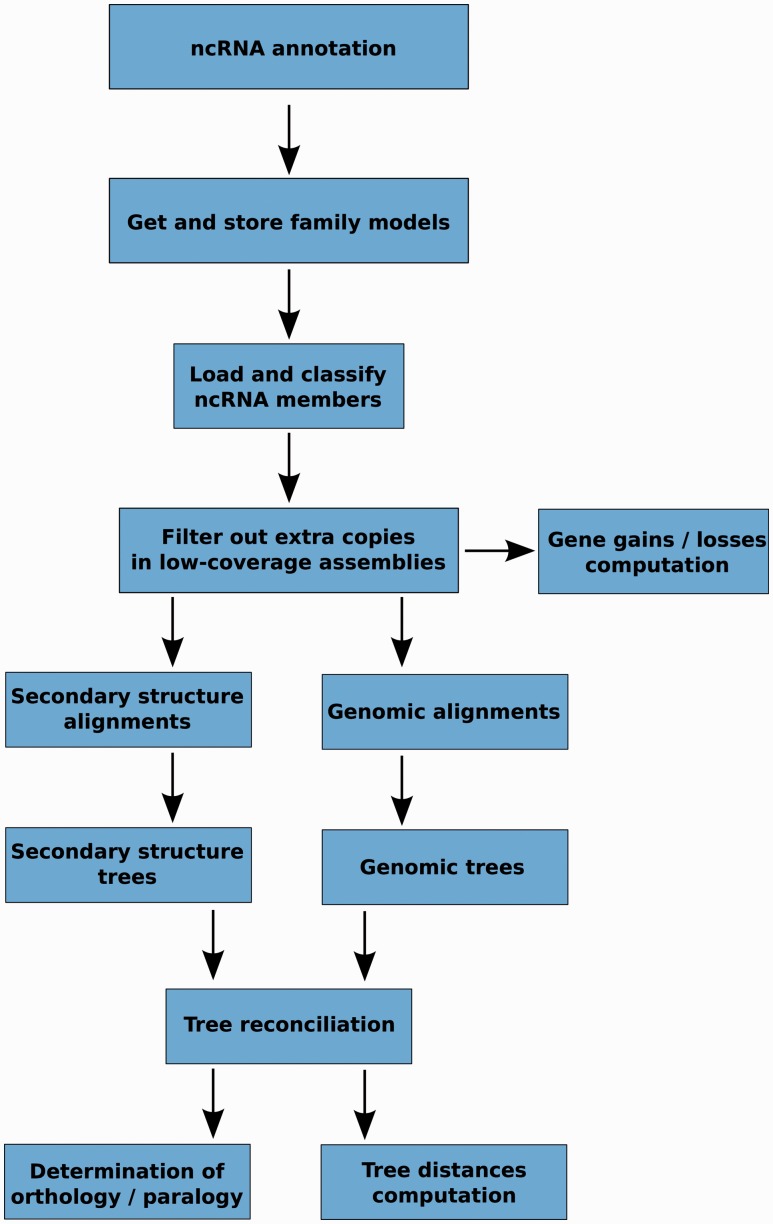
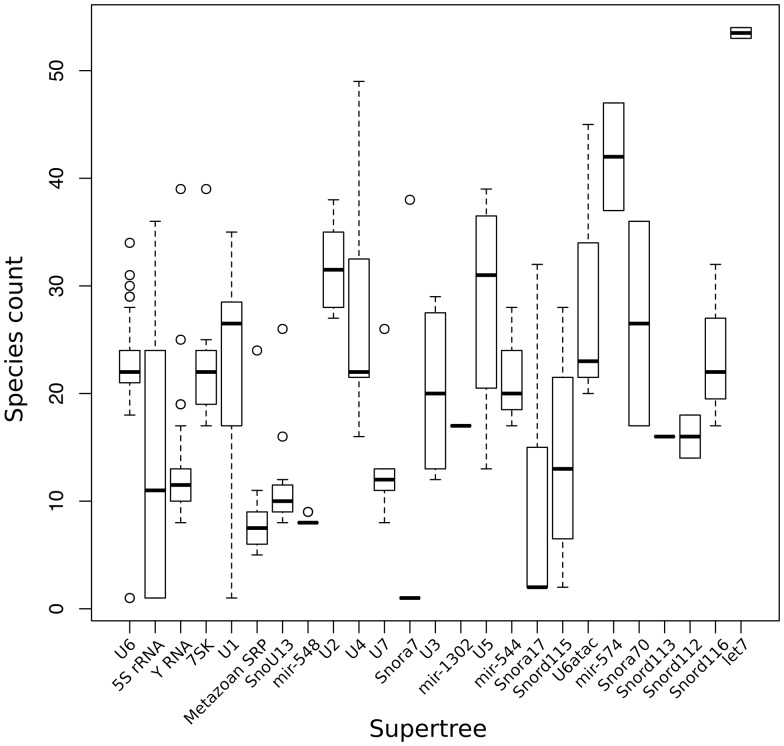
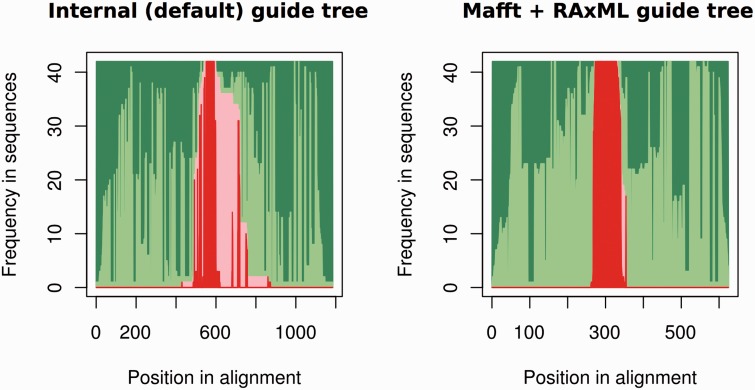
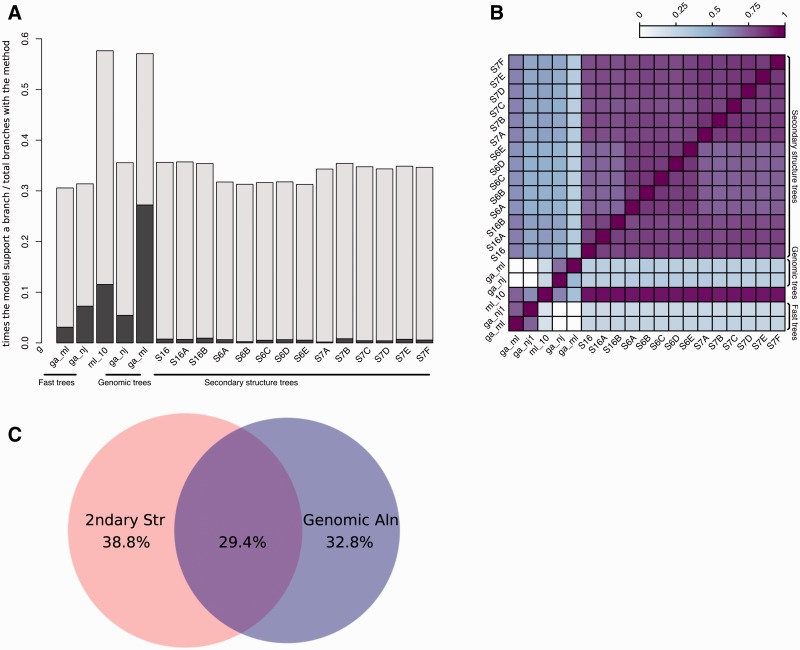
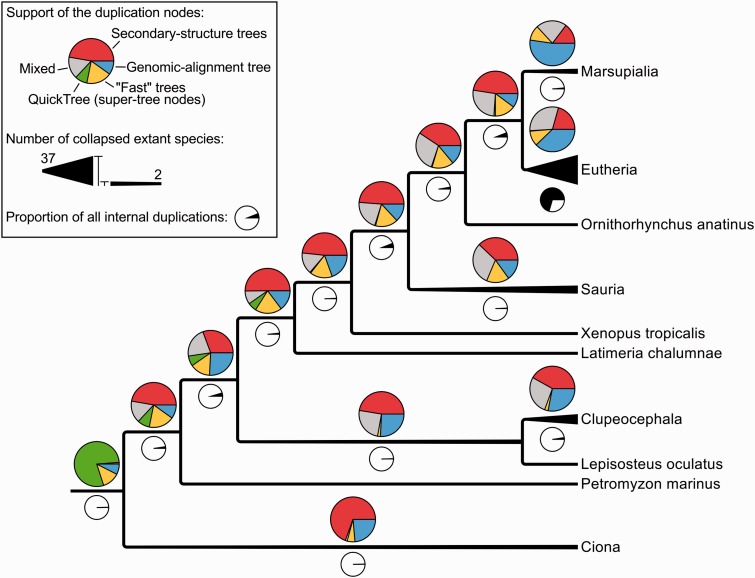
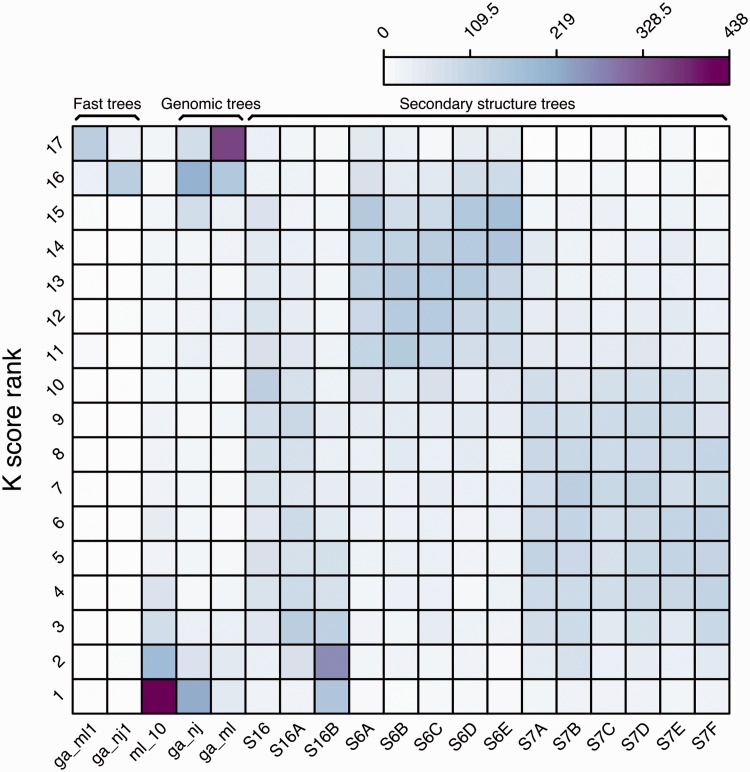
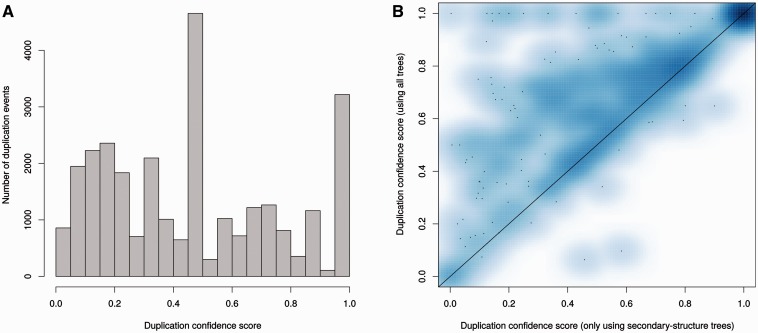
Annotation of orthologous and paralogous genes is necessary for many aspects of evolutionary analysis. Methods to infer these homology relationships have traditionally focused on protein-coding genes and evolutionary models used by these methods normally assume the positions in the protein evolve independently. However, as our appreciation for the roles of non-coding RNA genes has increased, consistently annotated sets of orthologous and paralogous ncRNA genes are increasingly needed. At the same time, methods such as PHASE or RAxML have implemented substitution models that consider pairs of sites to enable proper modelling of the loops and other features of RNA secondary structure. Here, we present a comprehensive analysis pipeline for the automatic detection of orthologues and paralogues for ncRNA genes. We focus on gene families represented in Rfam and for which a specific covariance model is provided. For each family ncRNA genes found in all Ensembl species are aligned using Infernal, and several trees are built using different substitution models. In parallel, a genomic alignment that includes the ncRNA genes and their flanking sequence regions is built with PRANK. This alignment is used to create two additional phylogenetic trees using the neighbour-joining (NJ) and maximum-likelihood (ML) methods. The trees arising from both the ncRNA and genomic alignments are merged using TreeBeST, which reconciles them with the species tree in order to identify speciation and duplication events. The final tree is used to infer the orthologues and paralogues following Fitch's definition. We also determine gene gain and loss events for each family using CAFE. All data are accessible through the Ensembl Comparative Genomics ('Compara') API, on our FTP site and are fully integrated in the Ensembl genome browser, where they can be accessed in a user-friendly manner. Database URL: http://www.ensembl.org.
© The Author(s) 2016. Published by Oxford University Press.
Figures
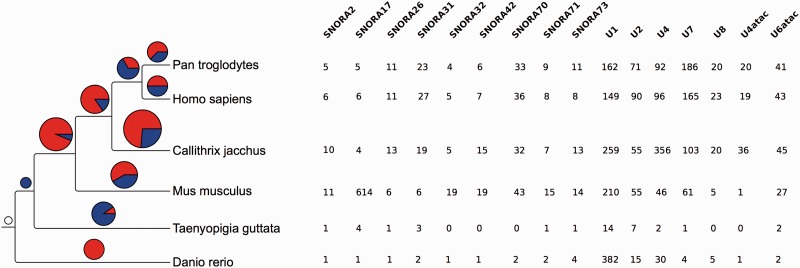
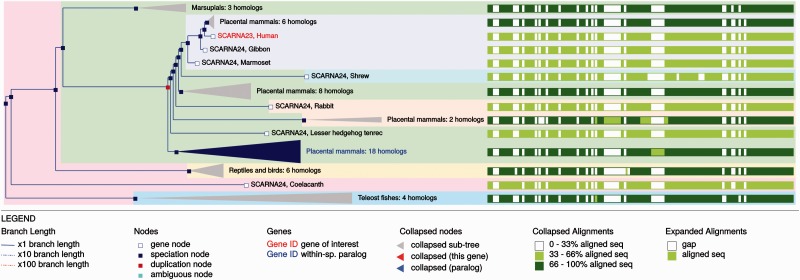
References
-
- Kishore S., Stamm S. (2006) The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science, 311, 230–232. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
