

FAK and paxillin, two potential targets in pancreatic cancer
- PMID: 26980710
- PMCID: PMC5058780
- DOI: 10.18632/oncotarget.8040
FAK and paxillin, two potential targets in pancreatic cancer
Abstract
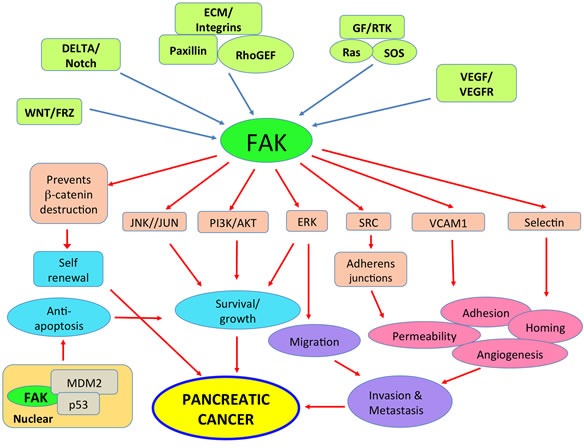
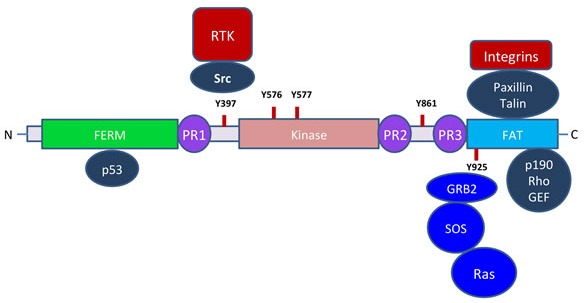
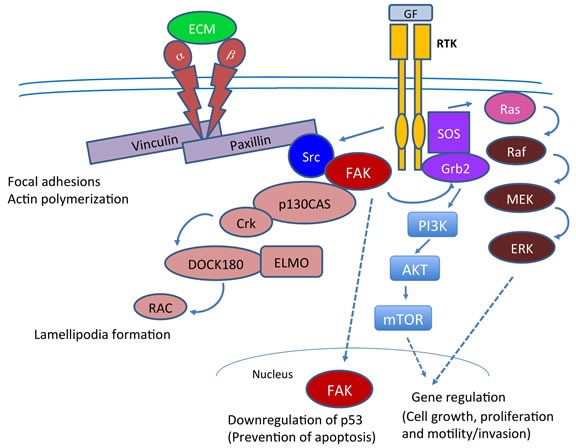
Pancreatic ductal adenocarcinoma (PDAC) is a devastating cancer in large part due to late diagnosis and a lack of effective screening tests. In spite of recent progress in imaging, surgery and new therapeutic options for pancreatic cancer, the overall five-year survival still remains unacceptably low. Numerous studies have shown that focal adhesion kinase (FAK) is activated in many cancers including PDAC and promotes cancer progression and metastasis. Paxillin, an intracellular adaptor protein that plays a key role in cytoskeletal organization, connects integrins to FAK and plays a key role in assembly and disassembly of focal adhesions. Here, we have reviewed evidence in support of FAK as a potential therapeutic target and summarized related combinatorial therapies.
Keywords: FAK; P53; integrins; pancreatic cancer; paxillin.
Conflict of interest statement
None.
Figures
Similar articles
-
Multi-targeted molecular therapeutic approach in aggressive neuroblastoma: the effect of Focal Adhesion Kinase-Src-Paxillin system.Expert Opin Ther Targets. 2014 Dec;18(12):1395-406. doi: 10.1517/14728222.2014.952280. Epub 2014 Sep 5. Expert Opin Ther Targets. 2014. PMID: 25189706 Review.
-
Targeted inhibition of CHKα and mTOR in models of pancreatic ductal adenocarcinoma: A novel regimen for metastasis.Cancer Lett. 2024 Nov 28;605:217280. doi: 10.1016/j.canlet.2024.217280. Epub 2024 Sep 28. Cancer Lett. 2024. PMID: 39343354
-
Focal adhesion kinase a potential therapeutic target for pancreatic cancer and malignant pleural mesothelioma.Cancer Biol Ther. 2018 Apr 3;19(4):316-327. doi: 10.1080/15384047.2017.1416937. Epub 2018 Feb 22. Cancer Biol Ther. 2018. PMID: 29303405 Free PMC article.
-
Targeting the Metastasis Suppressor, N-Myc Downstream Regulated Gene-1, with Novel Di-2-Pyridylketone Thiosemicarbazones: Suppression of Tumor Cell Migration and Cell-Collagen Adhesion by Inhibiting Focal Adhesion Kinase/Paxillin Signaling.Mol Pharmacol. 2016 May;89(5):521-40. doi: 10.1124/mol.115.103044. Epub 2016 Feb 19. Mol Pharmacol. 2016. PMID: 26895766 Free PMC article.
-
Exploring the therapeutic potential of focal adhesion kinase inhibition in overcoming chemoresistance in pancreatic ductal adenocarcinoma.Future Med Chem. 2024 Feb;16(3):271-289. doi: 10.4155/fmc-2023-0234. Epub 2024 Jan 25. Future Med Chem. 2024. PMID: 38269431 Review.
Cited by
-
The Role of CTHRC1 in Regulation of Multiple Signaling and Tumor Progression and Metastasis.Mediators Inflamm. 2020 Aug 12;2020:9578701. doi: 10.1155/2020/9578701. eCollection 2020. Mediators Inflamm. 2020. PMID: 32848510 Free PMC article. Review.
-
The 'Yin and Yang' of Cancer Cell Growth and Mechanosensing.Cancers (Basel). 2021 Sep 23;13(19):4754. doi: 10.3390/cancers13194754. Cancers (Basel). 2021. PMID: 34638240 Free PMC article. Review.
-
Norcantharidin Suppresses YD-15 Cell Invasion Through Inhibition of FAK/Paxillin and F-Actin Reorganization.Molecules. 2019 May 19;24(10):1928. doi: 10.3390/molecules24101928. Molecules. 2019. PMID: 31109130 Free PMC article.
-
Combination of Phenethyl Isothiocyanate and Dasatinib Inhibits Hepatocellular Carcinoma Metastatic Potential through FAK/STAT3/Cadherin Signalling and Reduction of VEGF Secretion.Pharmaceutics. 2023 Sep 27;15(10):2390. doi: 10.3390/pharmaceutics15102390. Pharmaceutics. 2023. PMID: 37896150 Free PMC article.
-
Paxillin: a crossroad in pathological cell migration.J Hematol Oncol. 2017 Feb 18;10(1):50. doi: 10.1186/s13045-017-0418-y. J Hematol Oncol. 2017. PMID: 28214467 Free PMC article. Review.
References
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA. 2015;65(1):5–29. - PubMed
-
- Hackert T, Buchler MW. Pancreatic cancer: advances in treatment, results and limitations. Digestive diseases. 2013;31(1):51–56. - PubMed
-
- Bilimoria KY, Bentrem DJ, Ko CY, Ritchey J, Stewart AK, Winchester DP, Talamonti MS. Validation of the 6th edition AJCC Pancreatic Cancer Staging System: report from the National Cancer Database. Cancer. 2007;110(4):738–744. - PubMed
-
- Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Molecular cancer therapeutics. 2007;6(4):1186–1197. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
