

Deletions linked to TP53 loss drive cancer through p53-independent mechanisms
- PMID: 26982726
- PMCID: PMC4836395
- DOI: 10.1038/nature17157
Deletions linked to TP53 loss drive cancer through p53-independent mechanisms
Abstract
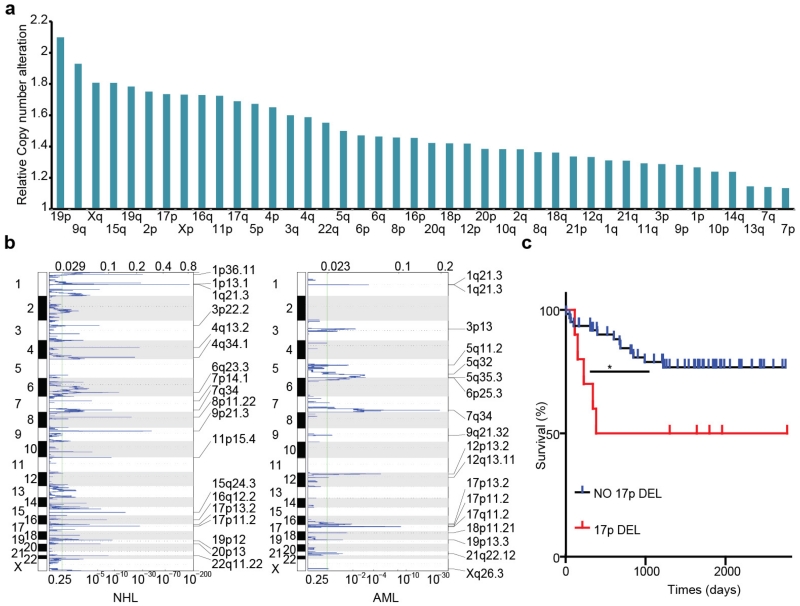
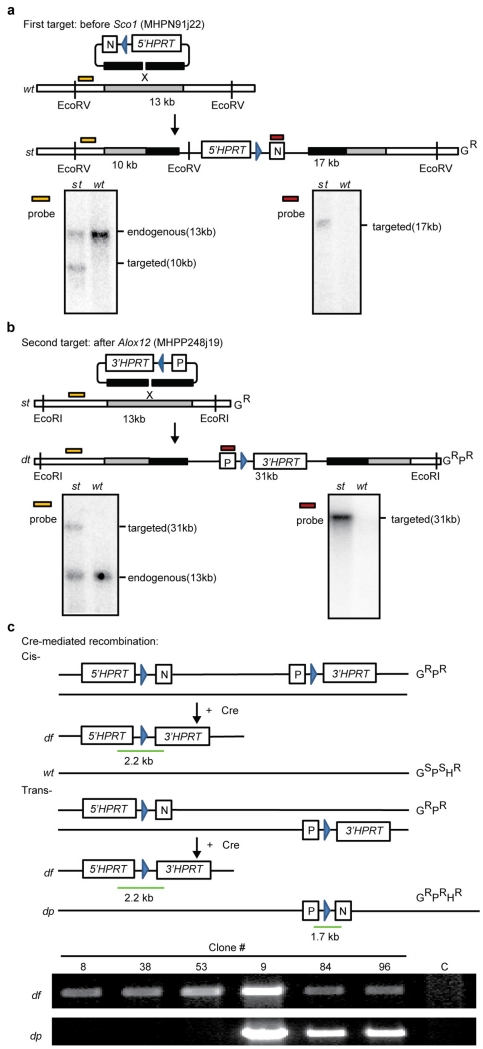
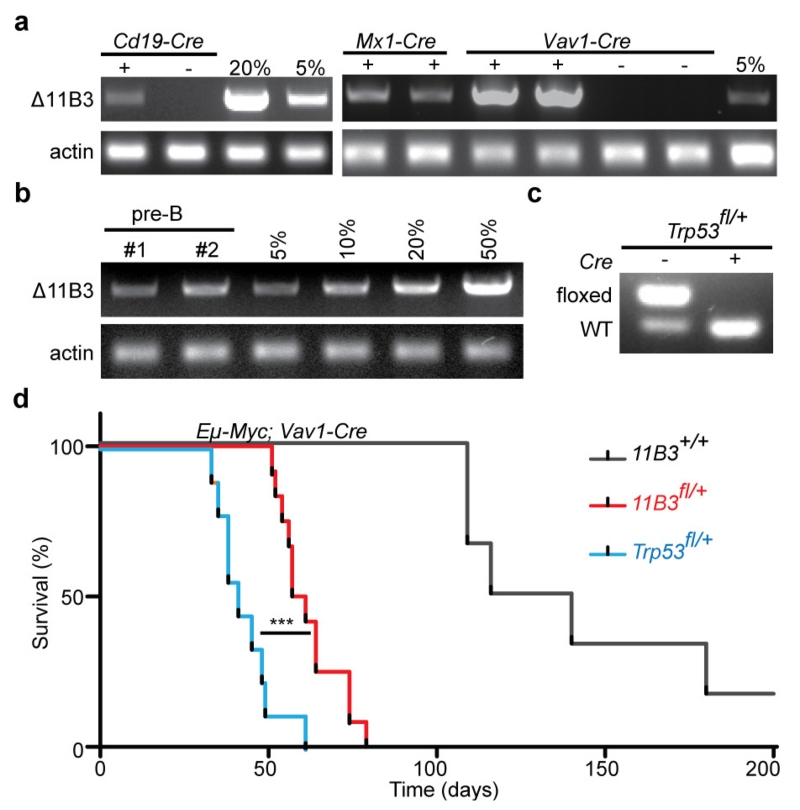
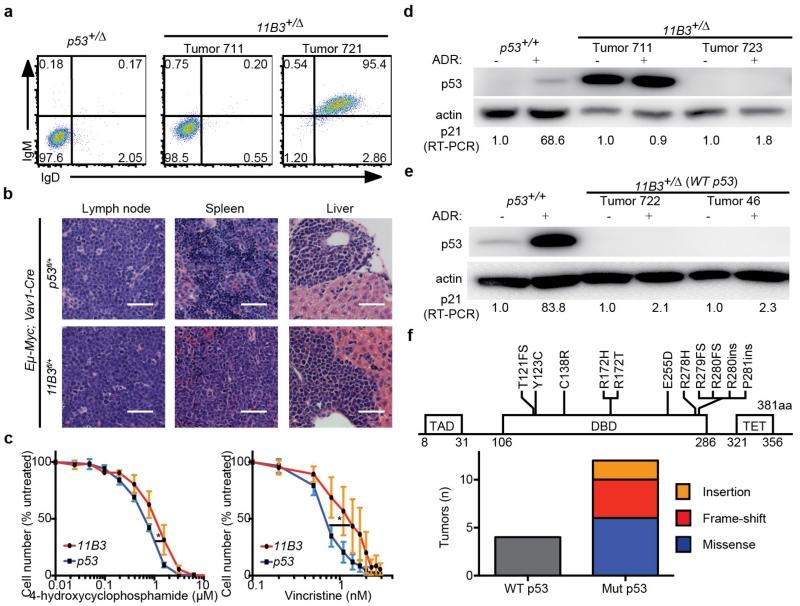
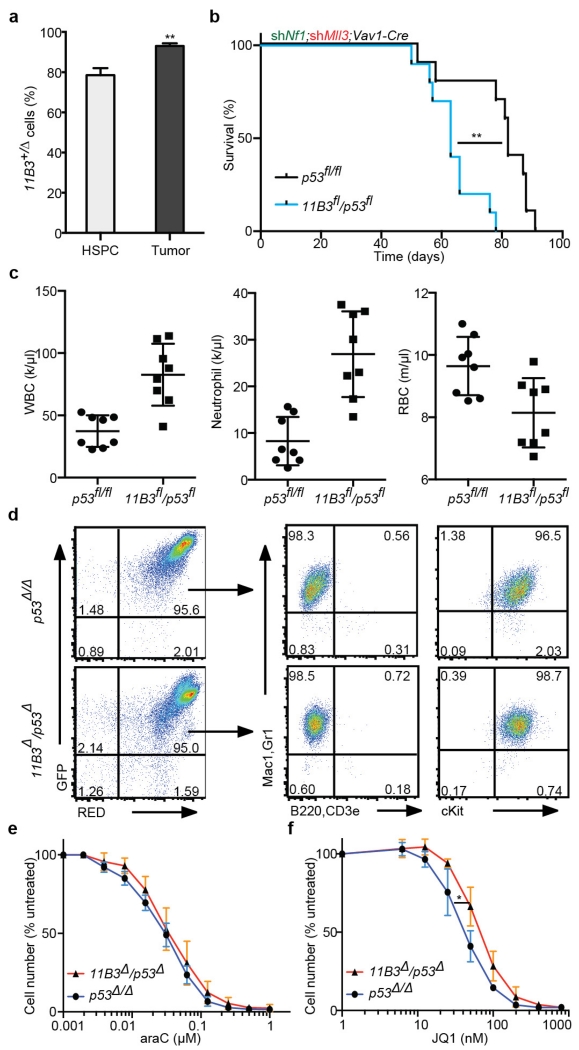
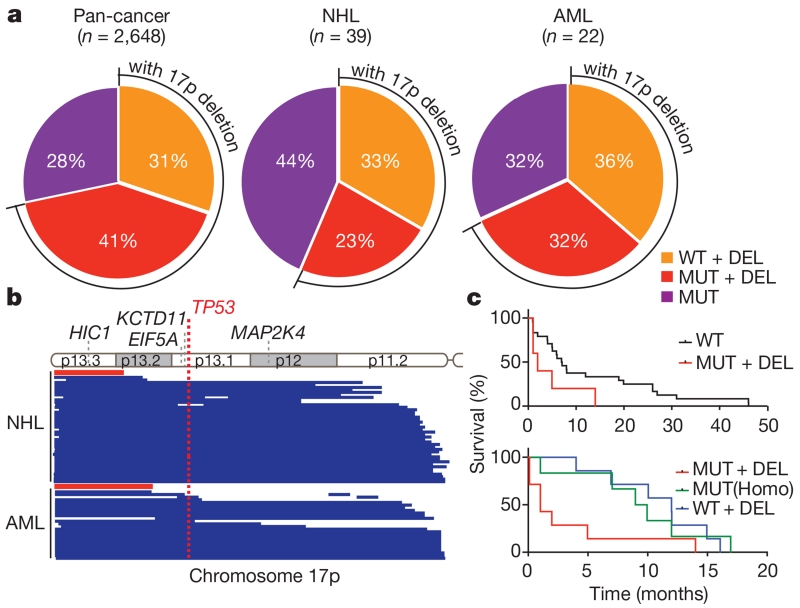
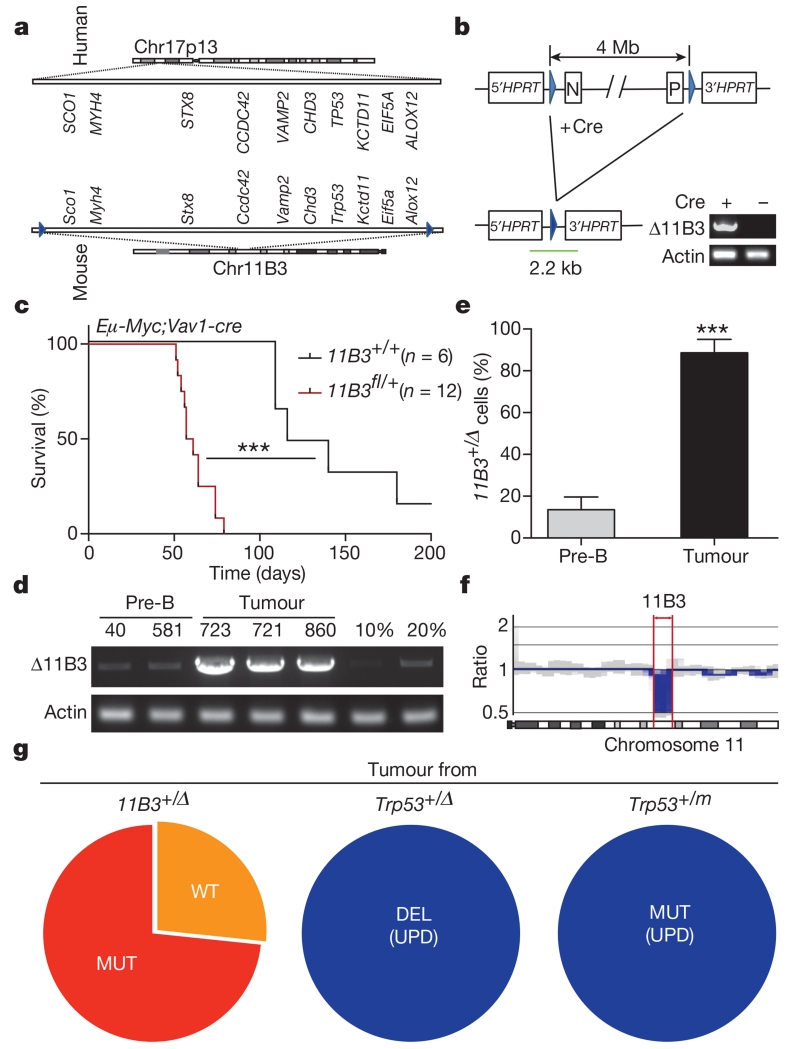
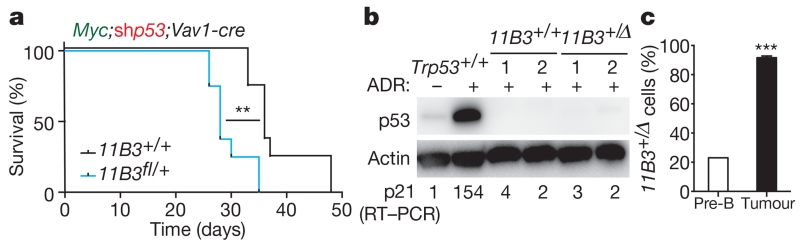
Mutations disabling the TP53 tumour suppressor gene represent the most frequent events in human cancer and typically occur through a two-hit mechanism involving a missense mutation in one allele and a 'loss of heterozygosity' deletion encompassing the other. While TP53 missense mutations can also contribute gain-of-function activities that impact tumour progression, it remains unclear whether the deletion event, which frequently includes many genes, impacts tumorigenesis beyond TP53 loss alone. Here we show that somatic heterozygous deletion of mouse chromosome 11B3, a 4-megabase region syntenic to human 17p13.1, produces a greater effect on lymphoma and leukaemia development than Trp53 deletion. Mechanistically, the effect of 11B3 loss on tumorigenesis involves co-deleted genes such as Eif5a and Alox15b (also known as Alox8), the suppression of which cooperates with Trp53 loss to produce more aggressive disease. Our results imply that the selective advantage produced by human chromosome 17p deletion reflects the combined impact of TP53 loss and the reduced dosage of linked tumour suppressor genes.
Figures
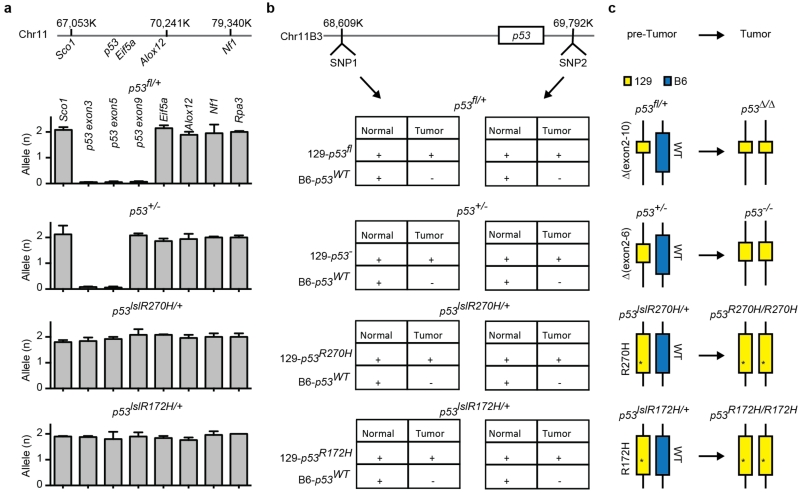
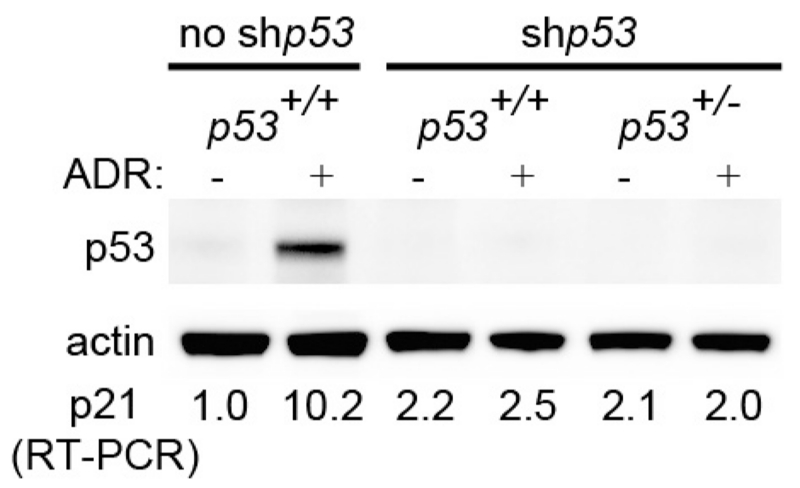
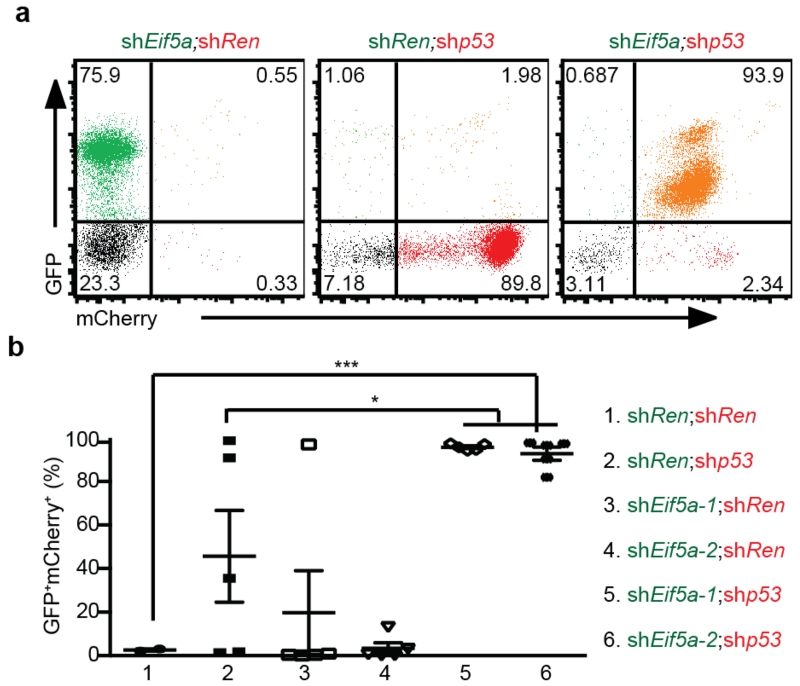
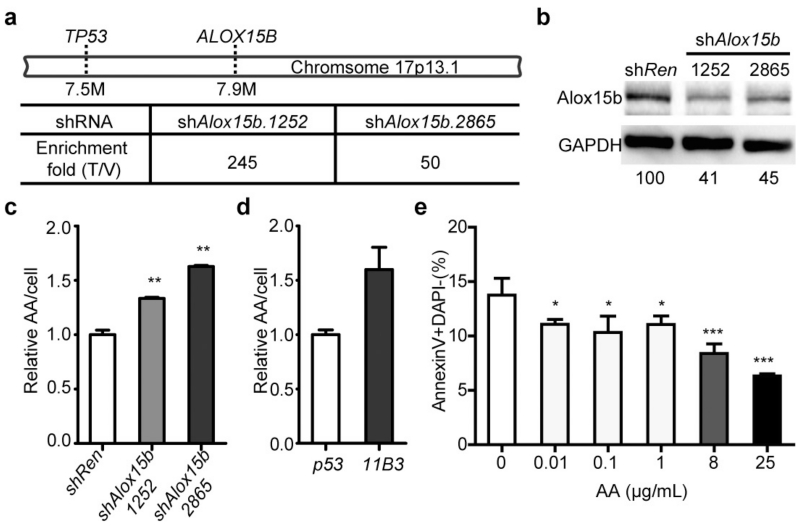
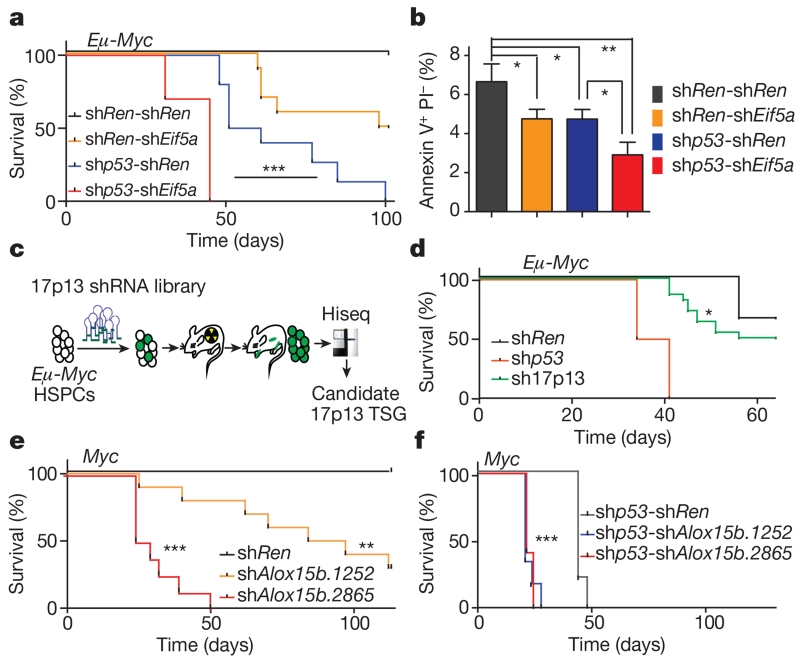
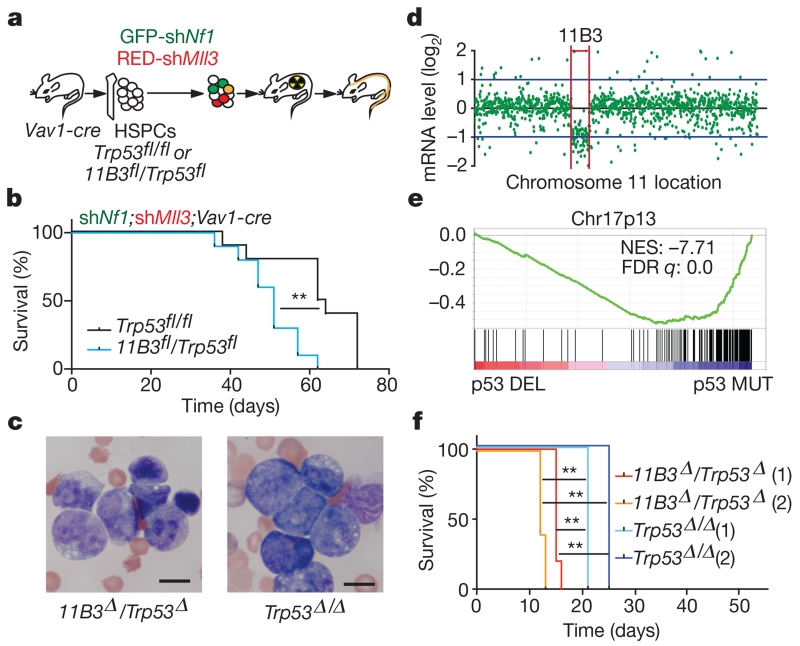
Comment in
-
TP53 Is Not the Only Driver of Chromosome 17p Loss.Cancer Discov. 2016 May;6(5):470. doi: 10.1158/2159-8290.CD-RW2016-058. Epub 2016 Mar 31. Cancer Discov. 2016. PMID: 27034379
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
