

Unifying Views of Autism Spectrum Disorders: A Consideration of Autoregulatory Feedback Loops
- PMID: 26985722
- PMCID: PMC5757244
- DOI: 10.1016/j.neuron.2016.02.017
Unifying Views of Autism Spectrum Disorders: A Consideration of Autoregulatory Feedback Loops
Abstract
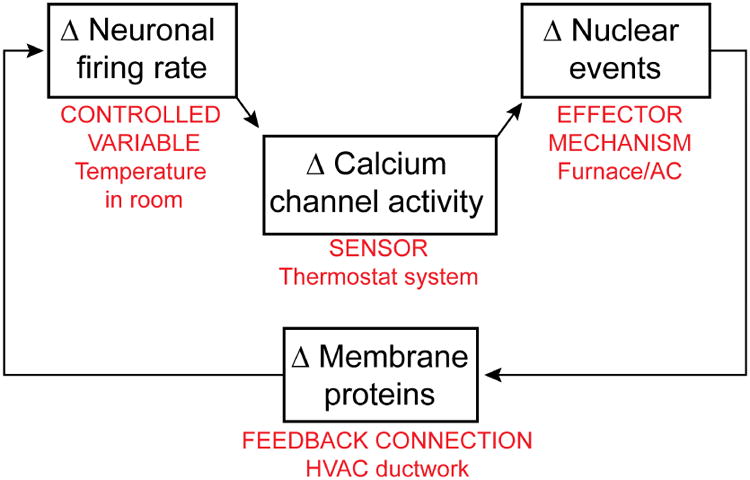
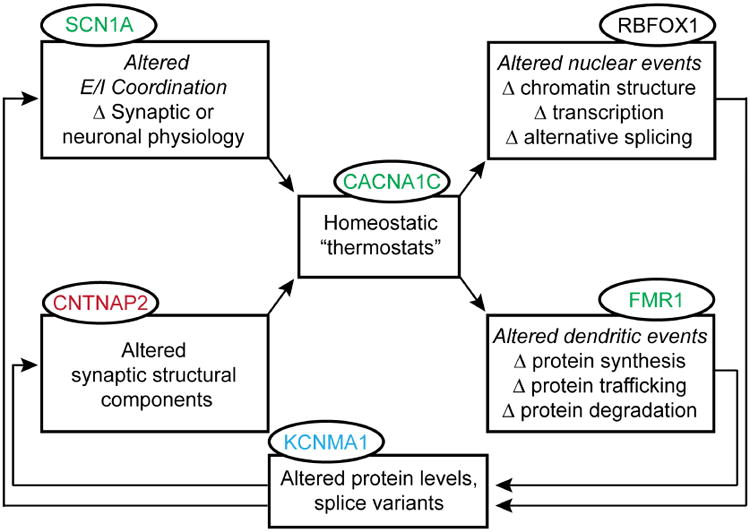
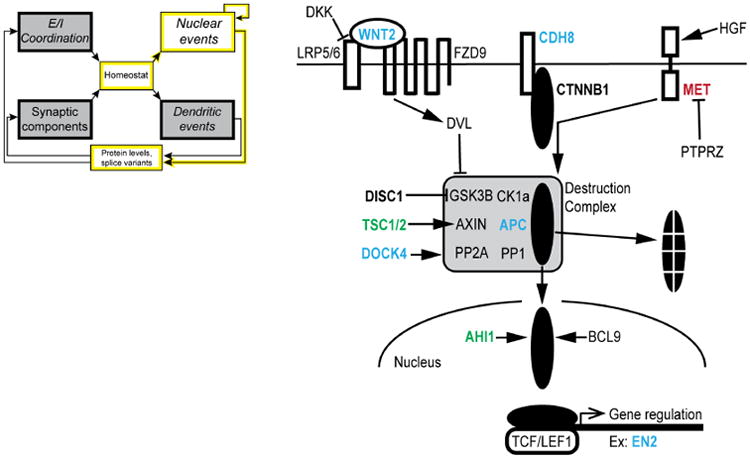
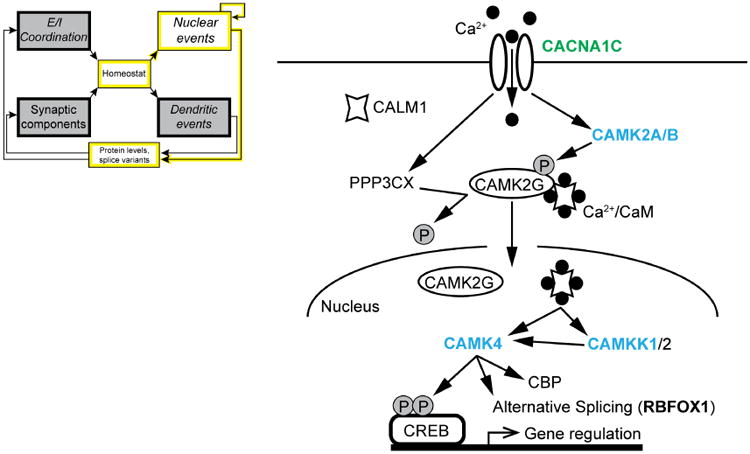
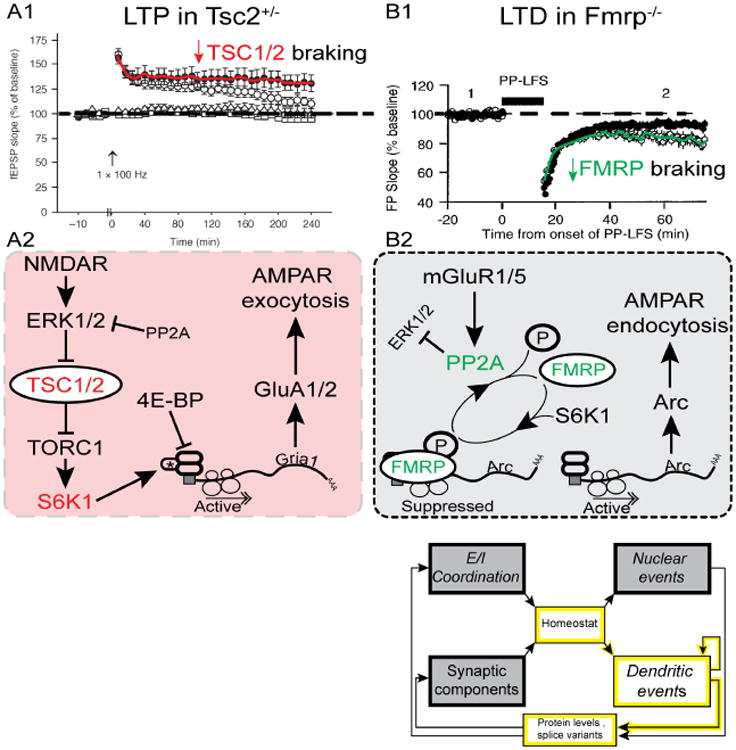
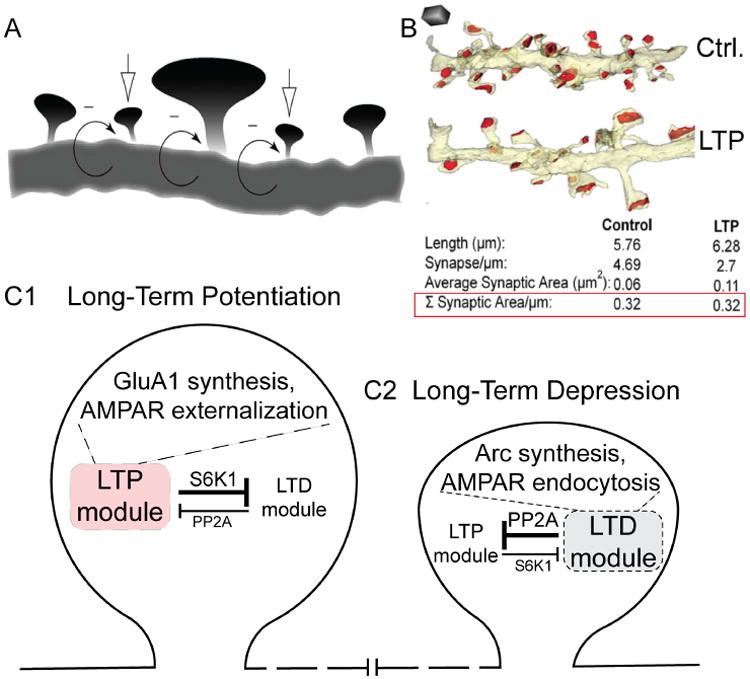
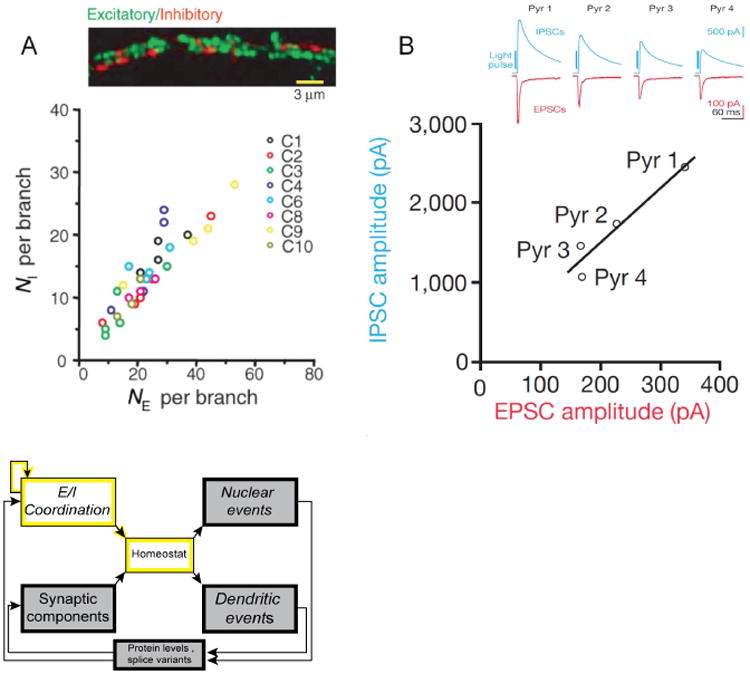
Understanding the mechanisms underlying autism spectrum disorders (ASDs) is a challenging goal. Here we review recent progress on several fronts, including genetics, proteomics, biochemistry, and electrophysiology, that raise motivation for forming a viable pathophysiological hypothesis. In place of a traditionally unidirectional progression, we put forward a framework that extends homeostatic hypotheses by explicitly emphasizing autoregulatory feedback loops and known synaptic biology. The regulated biological feature can be neuronal electrical activity, the collective strength of synapses onto a dendritic branch, the local concentration of a signaling molecule, or the relative strengths of synaptic excitation and inhibition. The sensor of the biological variable (which we have termed the homeostat) engages mechanisms that operate as negative feedback elements to keep the biological variable tightly confined. We categorize known ASD-associated gene products according to their roles in such feedback loops and provide detailed commentary for exemplar genes within each module.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
References
-
- Aguilar-Valles A, Matta-Camacho E, Khoutorsky A, Gkogkas C, Nader K, Lacaille JC, Sonenberg N. Inhibition of Group I Metabotropic Glutamate Receptors Reverses Autistic-Like Phenotypes Caused by Deficiency of the Translation Repressor eIF4E Binding Protein 2. J Neurosci. 2015;35:11125–11132. - PMC - PubMed
-
- Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P. Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr Biol. 1995;5:283–295. - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature genetics. 1999;23:185–188. - PubMed
-
- Anderson DJ, Jan YN. The determination of the neuronal phenotype. In: Cowan WM, Jessell TM, Zipursky SL, editors. Molecular and Cellular Approaches to Neural Development. New York: Oxford University Press; 1997. pp. 26–63.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
