

A Laboratory Phenotype/Genotype Correlation of 1167 French Patients From 670 Families With von Willebrand Disease: A New Epidemiologic Picture
- PMID: 26986123
- PMCID: PMC4839904
- DOI: 10.1097/MD.0000000000003038
A Laboratory Phenotype/Genotype Correlation of 1167 French Patients From 670 Families With von Willebrand Disease: A New Epidemiologic Picture
Abstract
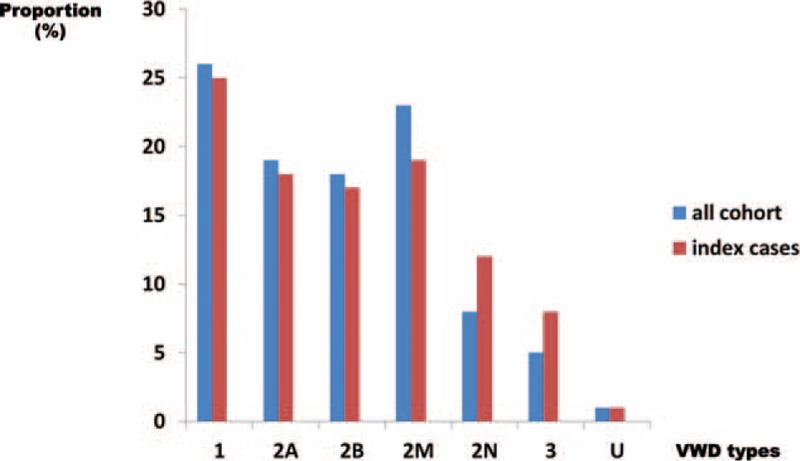
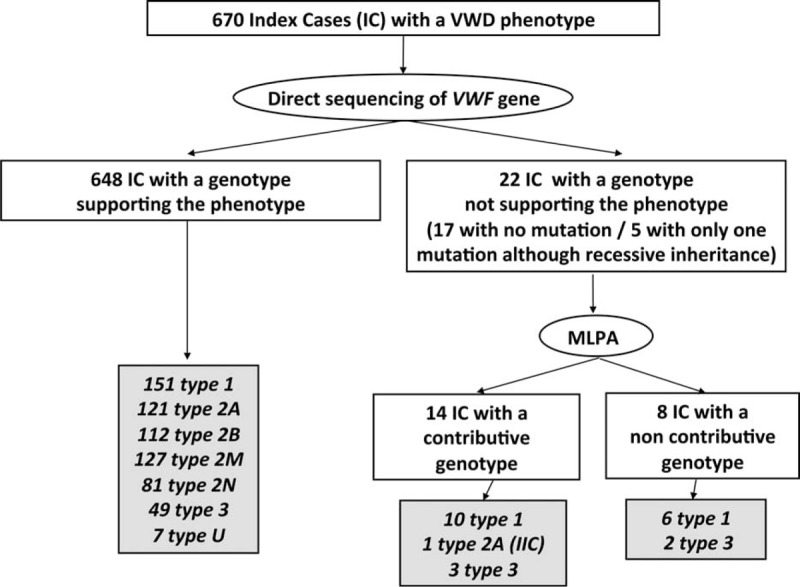
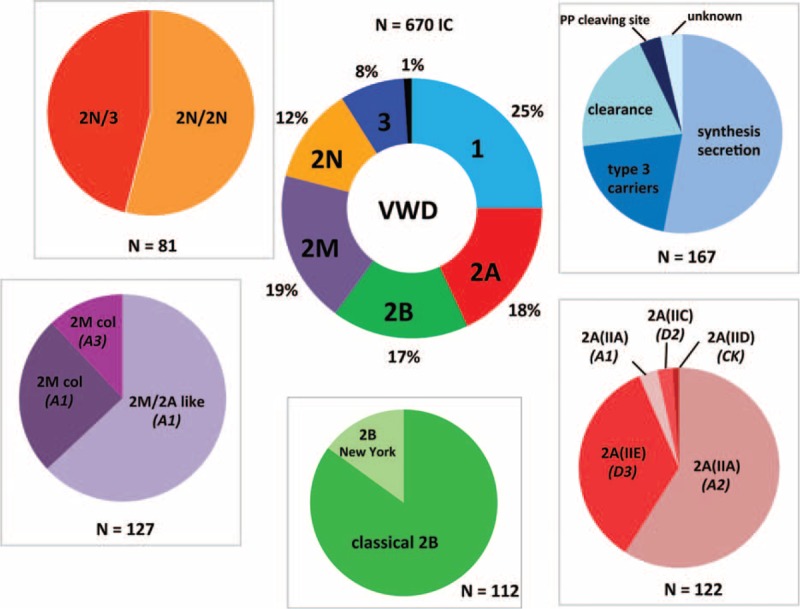
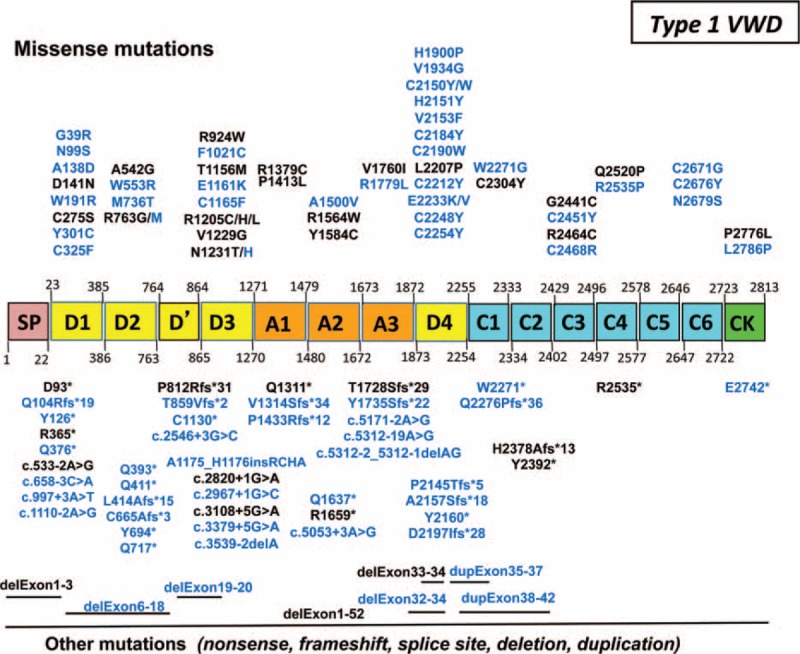
von Willebrand disease (VWD) is a genetic bleeding disease due to a defect of von Willebrand factor (VWF), a glycoprotein crucial for platelet adhesion to the subendothelium after vascular injury. VWD include quantitative defects of VWF, either partial (type 1 with VWF levels <50 IU/dL) or virtually total (type 3 with undetectable VWF levels) and also qualitative defects of VWF (type 2 variants with discrepant antigenic and functional VWF levels). The most bleeding forms of VWD usually do not concern type 1 patients with the mildest VWF defects (VWF levels between 30 and 50 IU/dL). The French reference center for VWD performed a laboratory phenotypic and genotypic analysis in 1167 VWD patients (670 families) selected by their basic biologic phenotype: type 3, type 2, and type 1 with VWF levels <30 IU/dL. In these patients indeed, to achieve an accurate diagnosis of VWD type and subtype is crucial for the management (treatment and genetic counseling). A phenotype/genotype correlation was present in 99.3% of cases; 323 distinct VWF sequence variations (58% of novel) were identified (missense 67% versus truncating 33%). The distribution of VWD types was: 25% of type 1, 8% of type 3, 66% of type 2 (2A: 18%, 2B: 17%, 2M: 19%, 2N: 12%), and 1% of undetermined type. Type 1 VWD was related either to a defective synthesis/secretion or to an accelerated clearance of VWF. In type 3 VWD, bi-allelic mutations of VWF were found in almost all patients. In type 2A, the most frequent mechanism was a hyper-proteolysis of VWF. Type 2B showed 85% of patients with deleterious mutations (distinct from type 2B New York). Type 2M was linked to a defective binding of VWF to platelet glycoprotein Ib or to collagen. Type 2N VWD included almost half type 2N/3. This biologic study emphasizes the complex mechanisms for both quantitative and qualitative VWF defects in VWD. In addition, this study provides a new epidemiologic picture of the most bleeding forms of VWD in which qualitative defects are predominant.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures
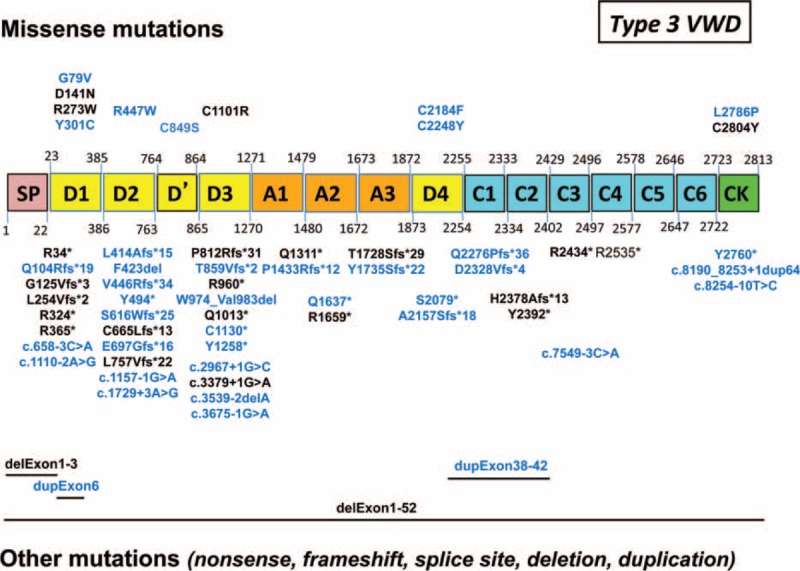
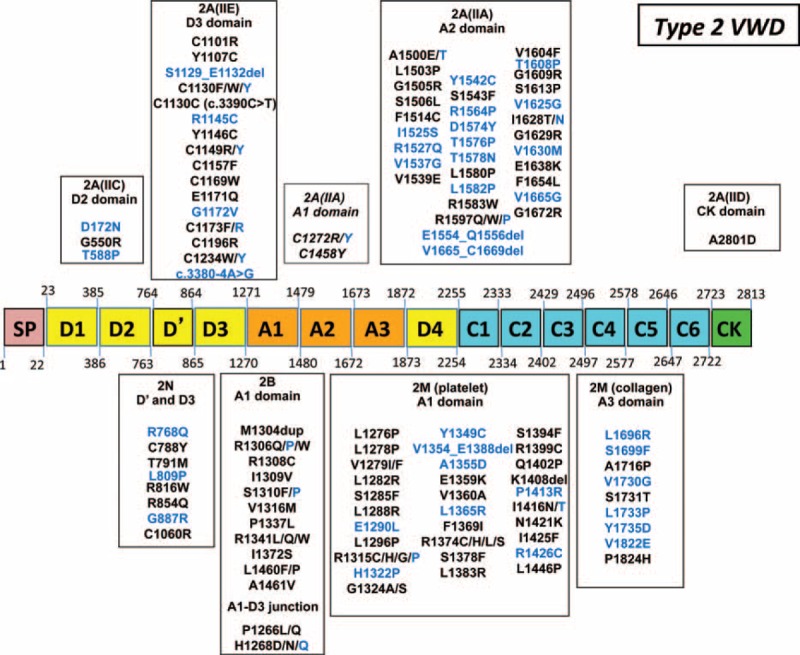
References
-
- Sadler JE. Von Willebrand factor in its native environment. Blood 2013; 121:2583–2584. - PubMed
-
- Lenting PJ, Christophe OD, Denis CV. Von Willebrand factor biosynthesis, secretion, and clearance: connecting the far ends. Blood 2015; 125:2019–2028. - PubMed
-
- Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev 2010; 24:123–134. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
