

The transcription factor scleraxis is a critical regulator of cardiac fibroblast phenotype
- PMID: 26988708
- PMCID: PMC4794909
- DOI: 10.1186/s12915-016-0243-8
The transcription factor scleraxis is a critical regulator of cardiac fibroblast phenotype
Abstract
Background: Resident fibroblasts synthesize the cardiac extracellular matrix, and can undergo phenotype conversion to myofibroblasts to augment matrix production, impairing function and contributing to organ failure. A significant gap in our understanding of the transcriptional regulation of these processes exists. Given the key role of this phenotype conversion in fibrotic disease, the identification of such novel transcriptional regulators may yield new targets for therapies for fibrosis.
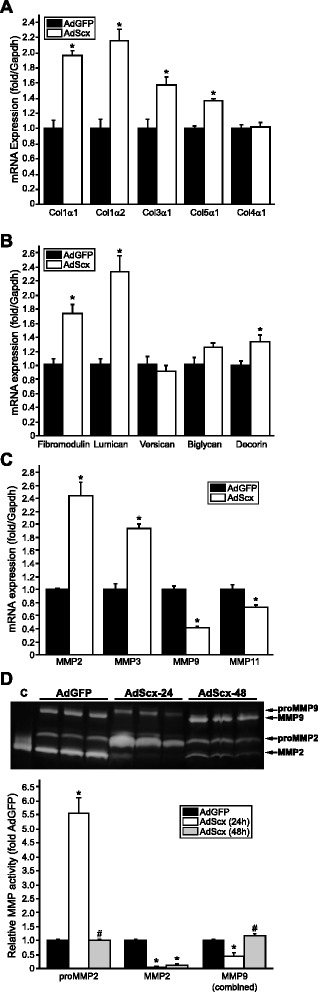
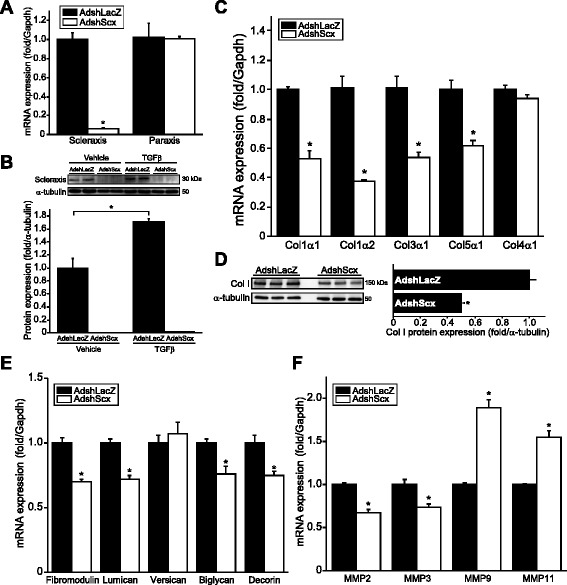
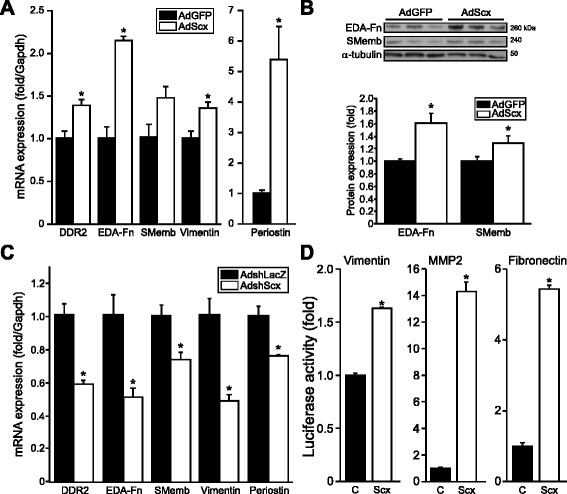
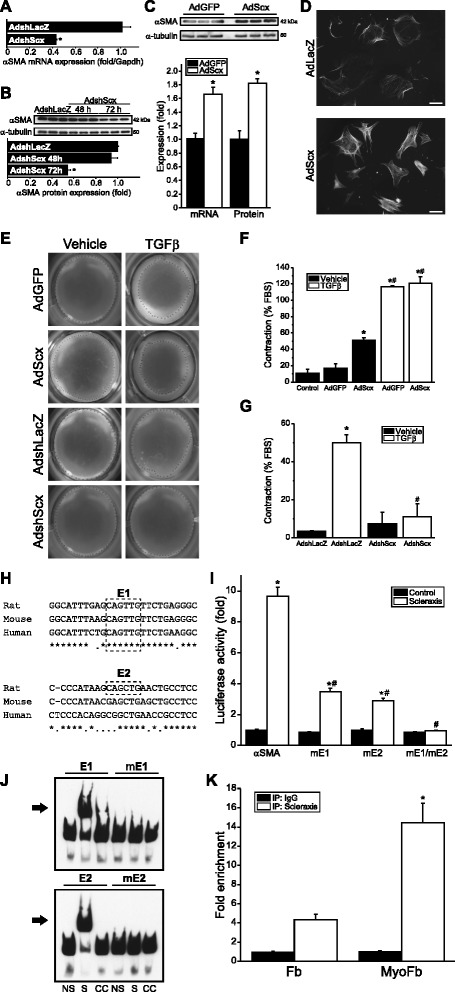
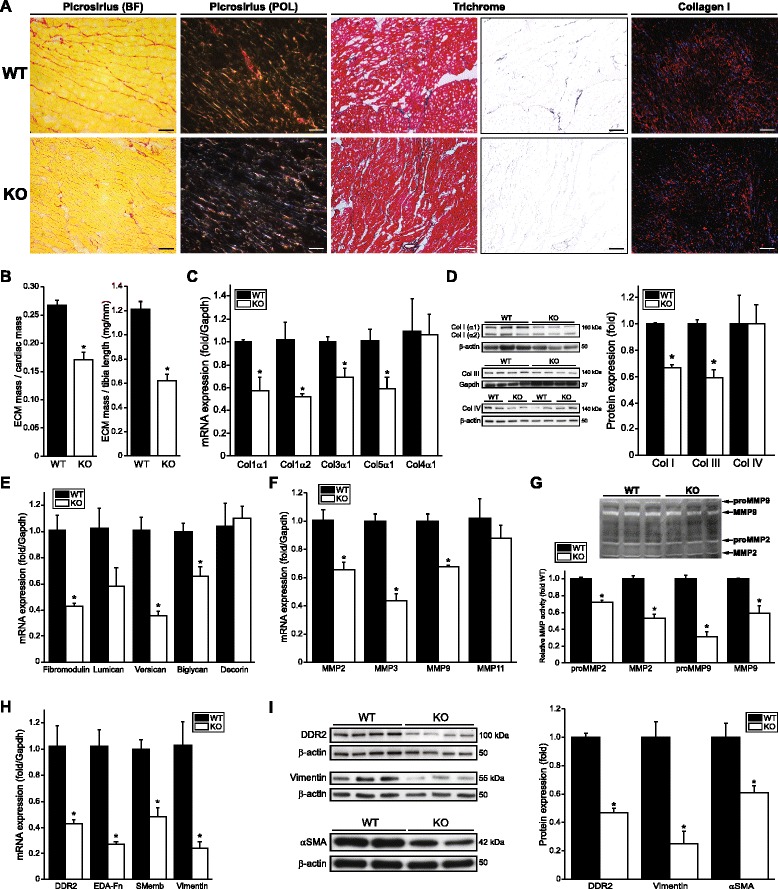
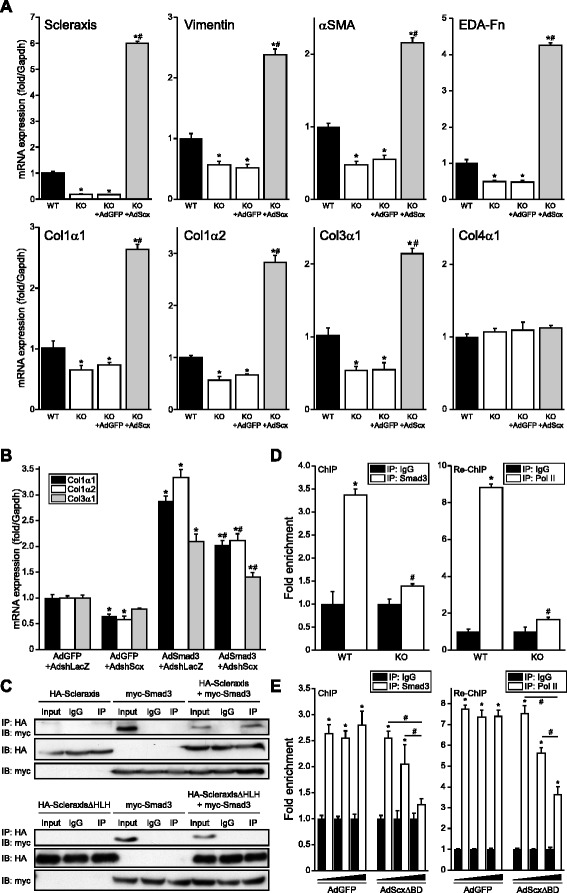
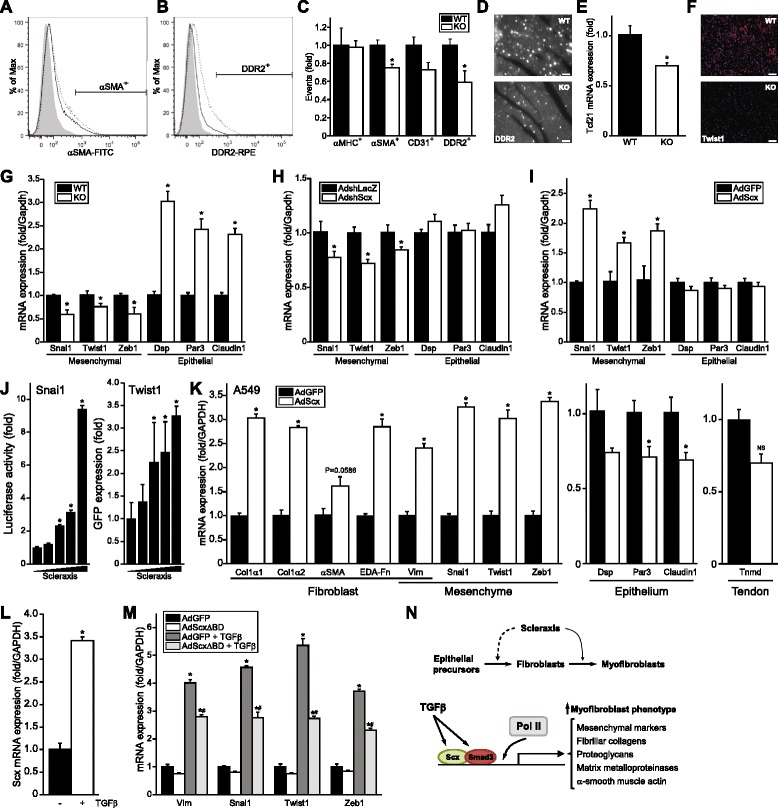
Results: Using explanted primary cardiac fibroblasts in gain- and loss-of-function studies, we found that scleraxis critically controls cardiac fibroblast/myofibroblast phenotype by direct transcriptional regulation of myriad genes that effectively define these cells, including extracellular matrix components and α-smooth muscle actin. Scleraxis furthermore potentiated the TGFβ/Smad3 signaling pathway, a key regulator of myofibroblast conversion, by facilitating transcription complex formation. While scleraxis promoted fibroblast to myofibroblast conversion, loss of scleraxis attenuated myofibroblast function and gene expression. These results were confirmed in scleraxis knockout mice, which were cardiac matrix-deficient and lost ~50% of their complement of cardiac fibroblasts, with evidence of impaired epithelial-to-mesenchymal transition (EMT). Scleraxis directly transactivated several EMT marker genes, and was sufficient to induce mesenchymal/fibroblast phenotype conversion of A549 epithelial cells. Conversely, loss of scleraxis attenuated TGFβ-induced EMT marker expression.
Conclusions: Our results demonstrate that scleraxis is a novel and potent regulator of cellular progression along the continuum culminating in the cardiac myofibroblast phenotype. Scleraxis was both sufficient to drive conversion, and required for full conversion to occur. Scleraxis fulfills this role by direct transcriptional regulation of key target genes, and by facilitating TGFβ/Smad signaling. Given the key role of fibroblast to myofibroblast conversion in fibrotic diseases in the heart and other tissue types, scleraxis may be an important target for therapeutic development.
Keywords: EMT; Extracellular matrix; Fibroblast; Gene expression; Myofibroblast; Phenoconversion; Transcription.
Figures
References
-
- Santiago JJ, Dangerfield AL, Rattan SG, Bathe KL, Cunnington RH, Raizman JE, et al. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn. 2010;239(6):1573–84. doi: 10.1002/dvdy.22280. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
