

Genome-Based Microbial Taxonomy Coming of Age
- PMID: 26988968
- PMCID: PMC4888819
- DOI: 10.1101/cshperspect.a018085
Genome-Based Microbial Taxonomy Coming of Age
Abstract
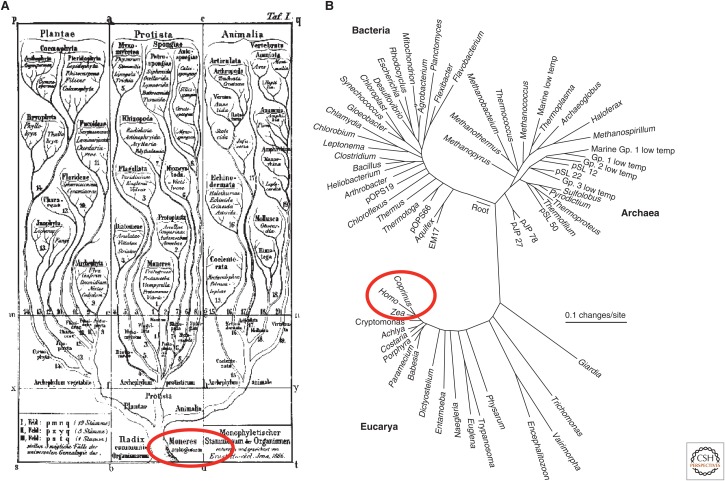
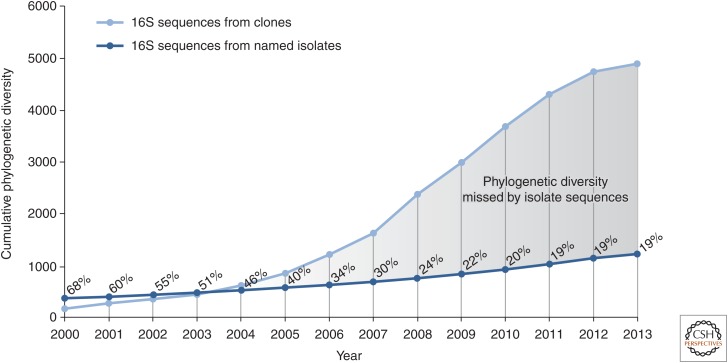
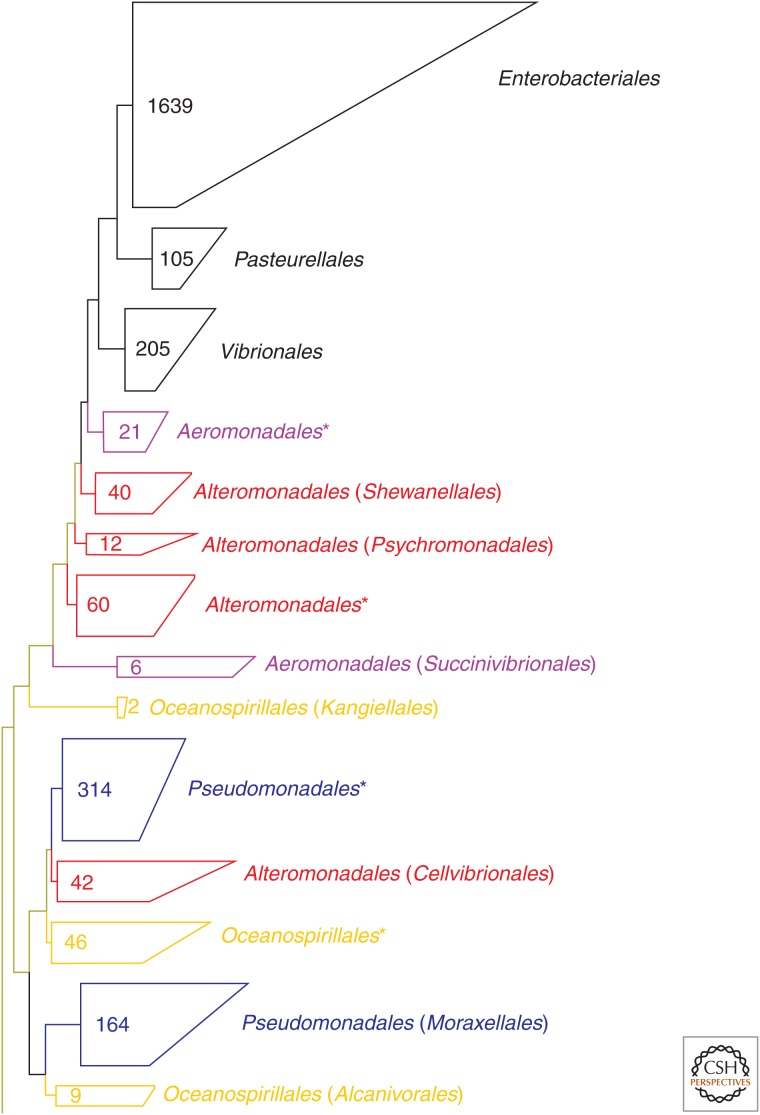
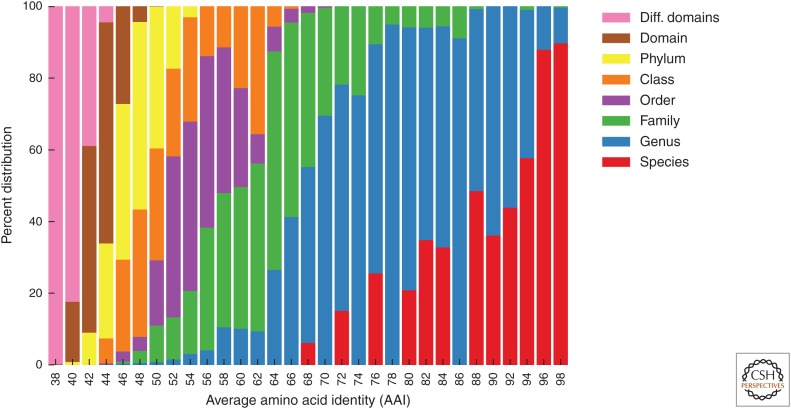
Reconstructing the complete evolutionary history of extant life on our planet will be one of the most fundamental accomplishments of scientific endeavor, akin to the completion of the periodic table, which revolutionized chemistry. The road to this goal is via comparative genomics because genomes are our most comprehensive and objective evolutionary documents. The genomes of plant and animal species have been systematically targeted over the past decade to provide coverage of the tree of life. However, multicellular organisms only emerged in the last 550 million years of more than three billion years of biological evolution and thus comprise a small fraction of total biological diversity. The bulk of biodiversity, both past and present, is microbial. We have only scratched the surface in our understanding of the microbial world, as most microorganisms cannot be readily grown in the laboratory and remain unknown to science. Ground-breaking, culture-independent molecular techniques developed over the past 30 years have opened the door to this so-called microbial dark matter with an accelerating momentum driven by exponential increases in sequencing capacity. We are on the verge of obtaining representative genomes across all life for the first time. However, historical use of morphology, biochemical properties, behavioral traits, and single-marker genes to infer organismal relationships mean that the existing highly incomplete tree is riddled with taxonomic errors. Concerted efforts are now needed to synthesize and integrate the burgeoning genomic data resources into a coherent universal tree of life and genome-based taxonomy.
Copyright © 2016 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538. - PubMed
-
- Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C. 2013. CONCOCT: Clustering cONtigs on COverage and ComposiTion. arXiv:1312.4038. - PubMed
-
- Amann R, Fuchs BM, Behrens S. 2001. The identification of microorganisms by fluorescence in situ hybridisation. Curr Opin Biotechnol 12: 231–236. - PubMed
-
- Arbogast BS, Edwards SV, Wakeley J, Beerli P, Slowinski JB. 2002. Estimating divergence times from molecular data on phylogenetic and population genetic timescales. Annu Rev Ecol Syst 33: 707–740.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous