

On the ability of molecular dynamics force fields to recapitulate NMR derived protein side chain order parameters
- PMID: 26990788
- PMCID: PMC4941777
- DOI: 10.1002/pro.2922
On the ability of molecular dynamics force fields to recapitulate NMR derived protein side chain order parameters
Abstract
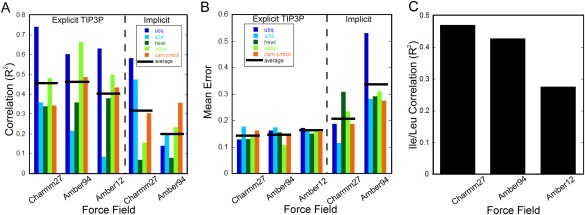
Molecular dynamics (MD) simulations have become a central tool for investigating various biophysical questions with atomistic detail. While many different proxies are used to qualify MD force fields, most are based on largely structural parameters such as the root mean square deviation from experimental coordinates or nuclear magnetic resonance (NMR) chemical shifts and residual dipolar couplings. NMR derived Lipari-Szabo squared generalized order parameter (O(2) ) values of amide NH bond vectors of the polypeptide chain were also often employed for refinement and validation. However, with a few exceptions, side chain methyl symmetry axis order parameters have not been incorporated into experimental reference sets. Using a test set of five diverse proteins, the performance of several force fields implemented in the NAMDD simulation package was examined. It was found that simulations employing explicit water implemented using the TIP3 model generally performed significantly better than those using implicit water in reproducing experimental methyl symmetry axis O(2) values. Overall the CHARMM27 force field performs nominally better than two implementations of the Amber force field. It appeared that recent quantum mechanics modifications to side chain torsional angles of leucine and isoleucine in the Amber force field have significantly hindered proper motional modeling for these residues. There remained significant room for improvement as even the best correlations of experimental and simulated methyl group Lipari-Szabo generalized order parameters fall below an R(2) of 0.8.
Keywords: NMR relaxation; accuracy; force field; molecular dynamics; protein motion; side chain motion.
© 2016 The Protein Society.
Figures
References
-
- Karplus M (2014) Development of multiscale models for complex chemical systems: from H+H‐2 to biomolecules. Angew Chem Intl Ed 53:9992–10005. - PubMed
-
- Levitt M (2014) Birth and future of multiscale modeling for macromolecular systems. Angew Chem Intl Ed 53:10006–10018. - PubMed
-
- Brooks BR, Brooks CL, 3rd , Mackerell AD, Jr. , Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30:154551. - PMC - PubMed
-
- Li D‐W, Brüschweiler R (2011) Iterative optimization of molecular mechanics force fields from NMR data of full‐length proteins. J Chem Theor Comp 7:1773–1782. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
