

Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon
- PMID: 26990986
- PMCID: PMC4889030
- DOI: 10.1016/j.molcel.2016.02.032
Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon
Abstract
Cereblon (CRBN), a substrate receptor for the cullin-RING ubiquitin ligase 4 (CRL4) complex, is a direct protein target for thalidomide teratogenicity and antitumor activity of immunomodulatory drugs (IMiDs). Here we report that glutamine synthetase (GS) is an endogenous substrate of CRL4(CRBN). Upon exposing cells to high glutamine concentration, GS is acetylated at lysines 11 and 14, yielding a degron that is necessary and sufficient for binding and ubiquitylation by CRL4(CRBN) and degradation by the proteasome. Binding of acetylated degron peptides to CRBN depends on an intact thalidomide-binding pocket but is not competitive with IMiDs. These findings reveal a feedback loop involving CRL4(CRBN) that adjusts GS protein levels in response to glutamine and uncover a new function for lysine acetylation.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures



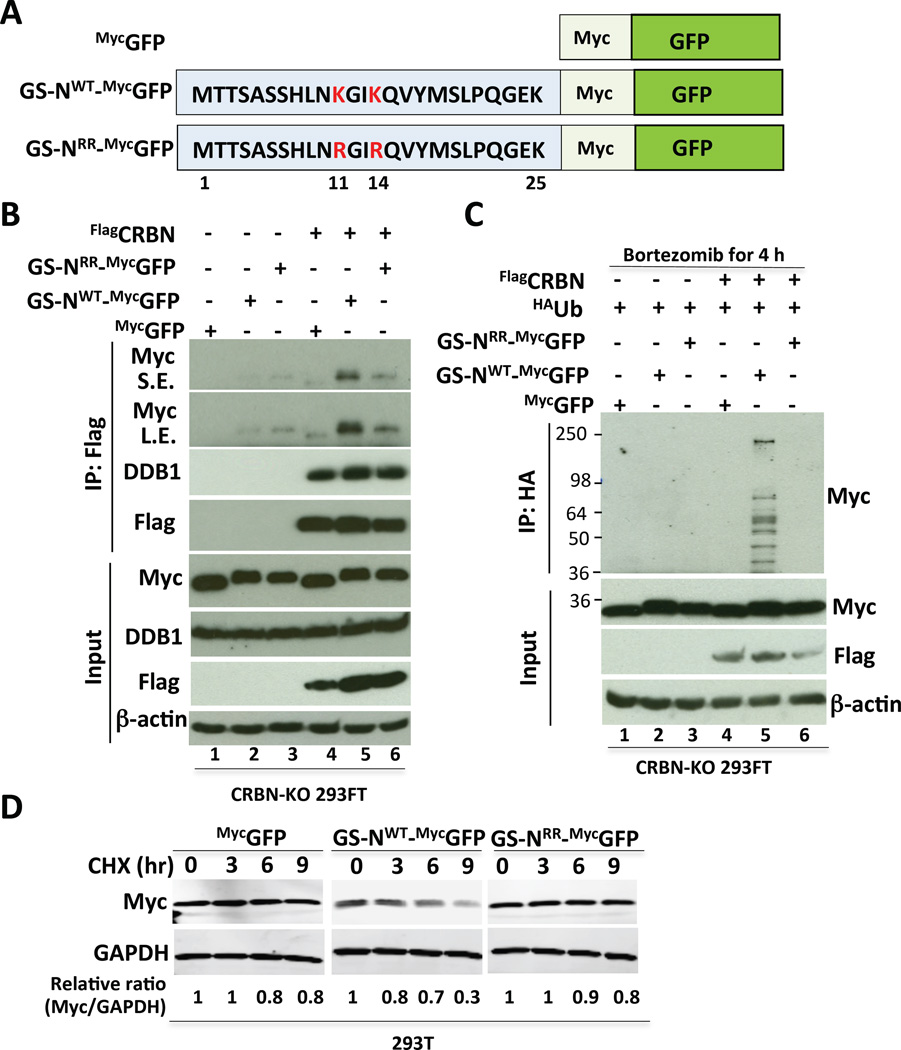

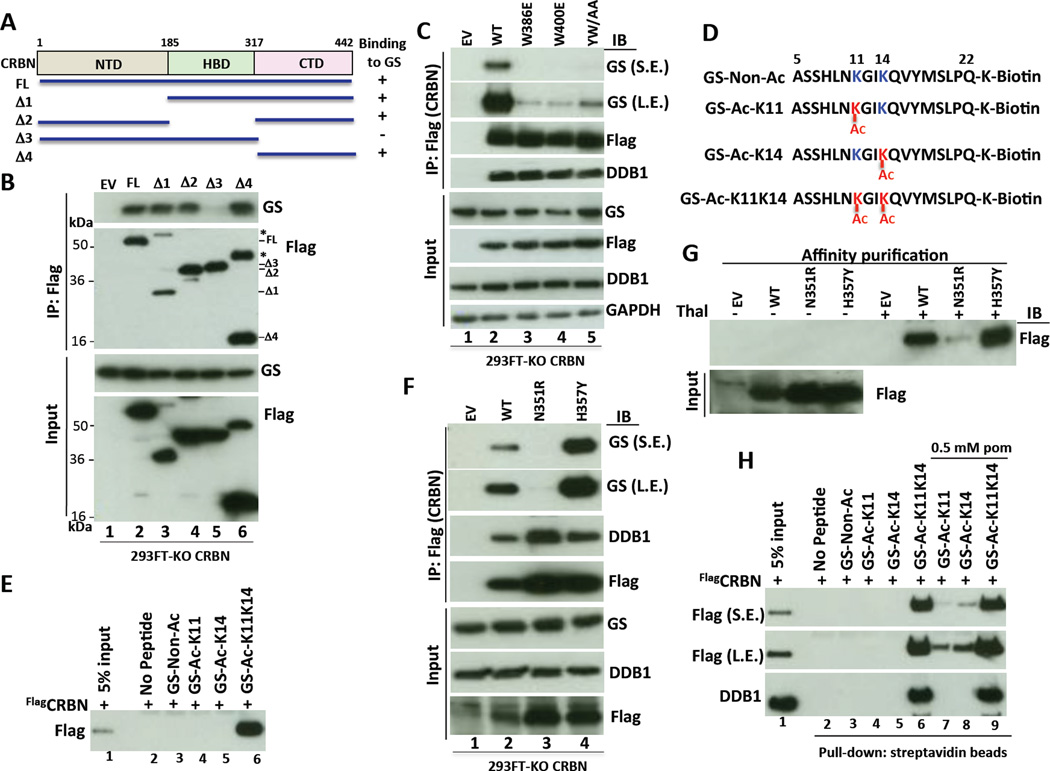
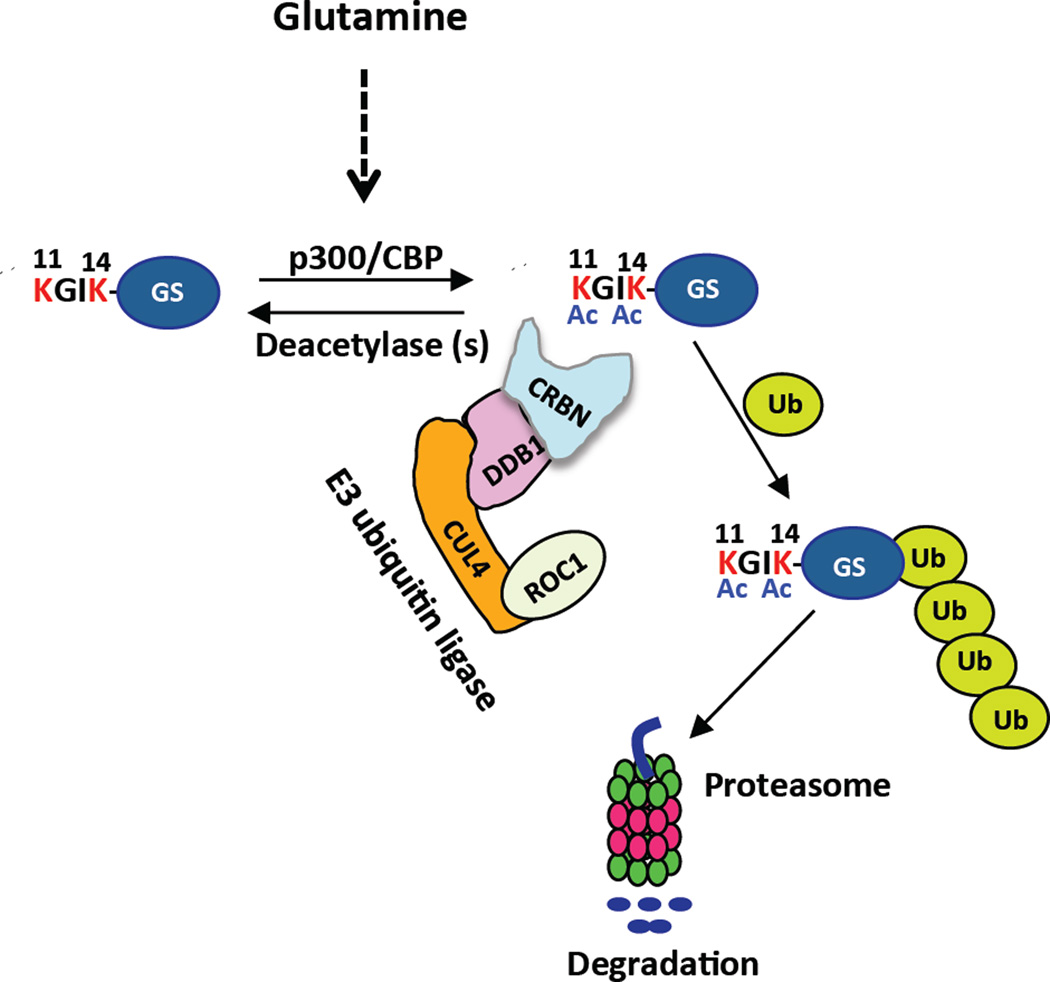
Comment in
-
An Acetyldegron Triggers CRBN to Take Down the "Q".Mol Cell. 2016 Mar 17;61(6):795-6. doi: 10.1016/j.molcel.2016.03.003. Mol Cell. 2016. PMID: 26990985
References
-
- Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. - PubMed
-
- Arad G, Freikopf A, Kulka RG. Glutamine-stimulated modification and degradation of glutamine synthetase in hepatoma tissue culture cells. Cell. 1976;8:95–101. - PubMed
-
- Araki T, Nagarajan R, Milbrandt J. Identification of genes induced in peripheral nerve after injury. Expression profiling and novel gene discovery. The Journal of biological chemistry. 2001;276:34131–34141. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
