

Soluble Adenylyl Cyclase Regulates Bile Salt-Induced Apoptosis in Human Cholangiocytes
- PMID: 26991014
- PMCID: PMC5111777
- DOI: 10.1002/hep.28550
Soluble Adenylyl Cyclase Regulates Bile Salt-Induced Apoptosis in Human Cholangiocytes
Abstract
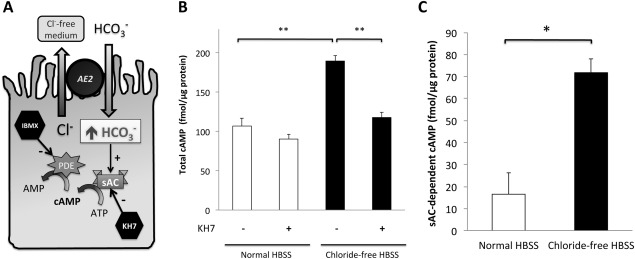
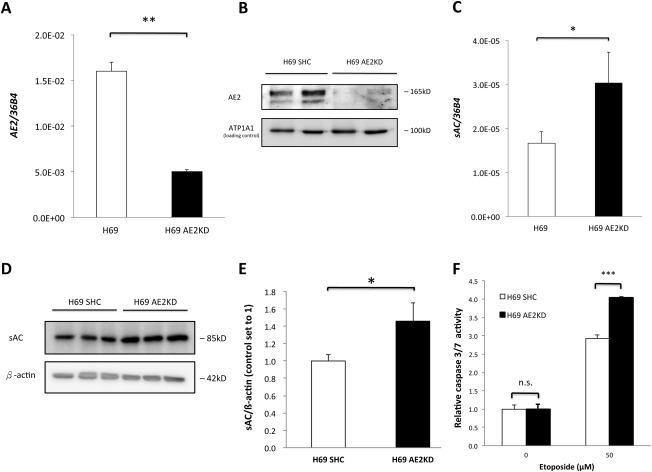
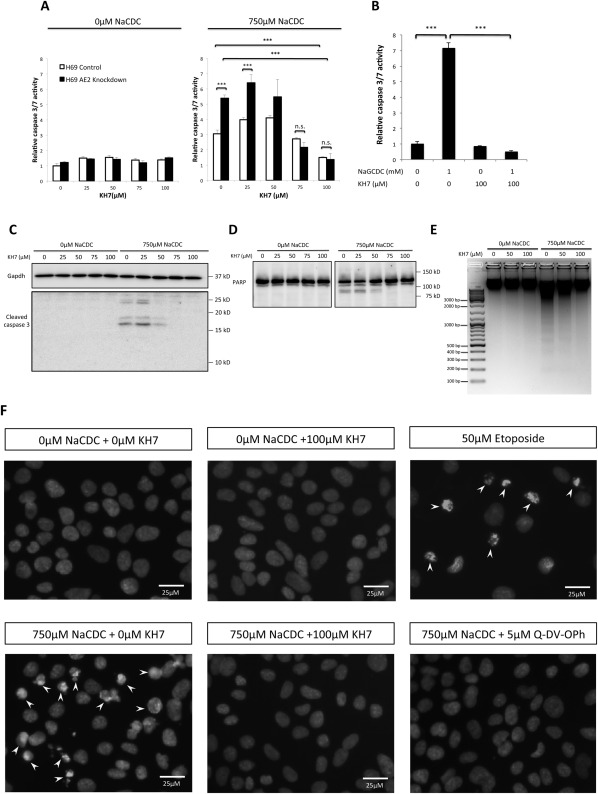
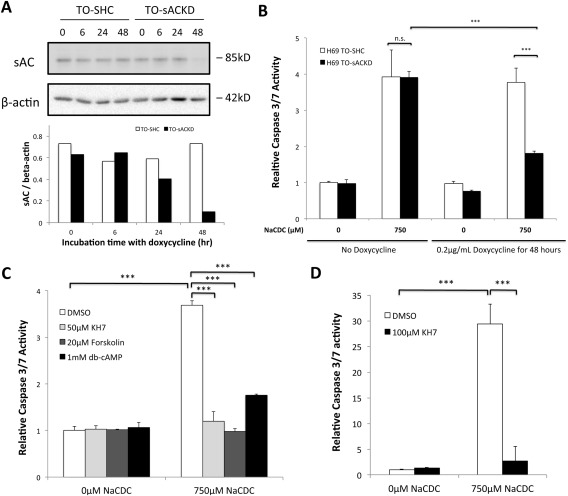
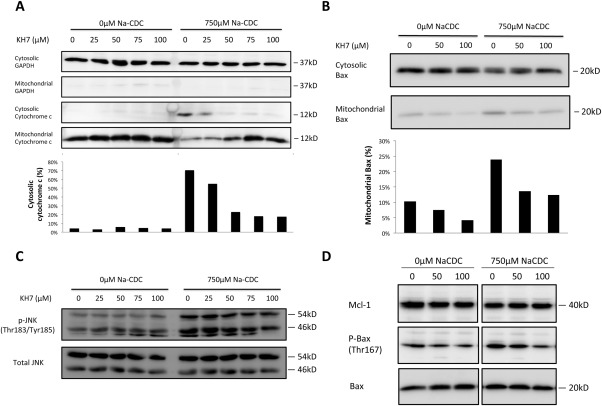
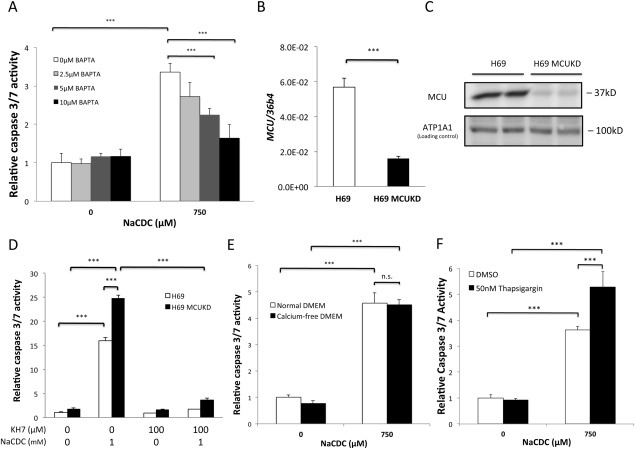
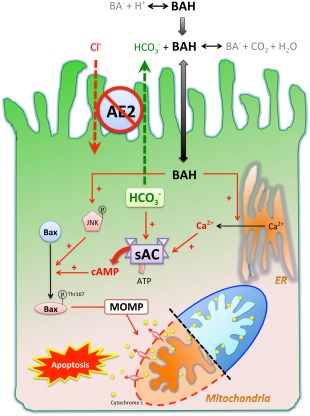
Anion exchanger 2 (AE2), the principal bicarbonate secretor in the human biliary tree, is down-regulated in primary biliary cholangitis. AE2 creates a "bicarbonate umbrella" that protects cholangiocytes from the proapoptotic effects of bile salts by maintaining them deprotonated. We observed that knockdown of AE2 sensitized immortalized H69 human cholangiocytes to not only bile salt-induced apoptosis (BSIA) but also etoposide-induced apoptosis. Because the toxicity of etoposide is pH-independent, there could be a more general mechanism for sensitization of AE2-depleted cholangiocytes to apoptotic stimuli. We found that AE2 deficiency led to intracellular bicarbonate accumulation and increased expression and activity of soluble adenylyl cyclase (sAC), an evolutionarily conserved bicarbonate sensor. Thus, we hypothesized that sAC regulates BSIA. H69 cholangiocytes and primary mouse cholangiocytes were used as models. The sAC-specific inhibitor KH7 not only reversed sensitization to BSIA in AE2-depleted H69 cholangiocytes but even completely prevented BSIA. sAC knockdown by tetracycline-inducible short hairpin RNA also prevented BSIA. In addition, sAC inhibition reversed BSIA membrane blebbing, nuclear condensation, and DNA fragmentation. Furthermore, sAC inhibition also prevented BSIA in primary mouse cholangiocytes. Mechanistically, sAC inhibition prevented Bax phosphorylation at Thr167 and mitochondrial translocation of Bax and cytochrome c release but not c-Jun N-terminal kinase activation during BSIA. Finally, BSIA in H69 cholangiocytes was inhibited by intracellular Ca(2+) chelation, aggravated by thapsigargin, and unaffected by removal of extracellular calcium.
Conclusions: BSIA is regulated by sAC, depends on intracellular Ca(2+) stores, and is mediated by the intrinsic apoptotic pathway; down-regulation of AE2 in primary biliary cholangitis sensitizes cholangiocytes to apoptotic insults by activating sAC, which may play a crucial role in disease pathogenesis. (Hepatology 2016;64:522-534).
Copyright © 2016 The Authors. HEPATOLOGY published by Wiley Periodicals, Inc., on behalf of the American Association for the Study of Liver Diseases.
Figures
Comment in
-
New evidence supporting the biliary bicarbonate umbrella theory.Clin Res Hepatol Gastroenterol. 2017 Mar;41(2):126-128. doi: 10.1016/j.clinre.2016.09.004. Epub 2016 Nov 3. Clin Res Hepatol Gastroenterol. 2017. PMID: 27818187 No abstract available.
References
-
- Selmi C, Bowlus CL, Gershwin ME, Coppel RL. Primary biliary cirrhosis. Lancet 2011;377:1600–1609. - PubMed
-
- Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology 2004;127:485–492. - PubMed
-
- Medina JF, Martinez A, Vazquez JJ, Prieto J. Decreased anion exchanger 2 immunoreactivity in the liver of patients with primary biliary cirrhosis. Hepatology 1997;25:12–17. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
