

Molecular Control of Atypical Protein Kinase C: Tipping the Balance between Self-Renewal and Differentiation
- PMID: 26992354
- PMCID: PMC4848065
- DOI: 10.1016/j.jmb.2016.03.003
Molecular Control of Atypical Protein Kinase C: Tipping the Balance between Self-Renewal and Differentiation
Abstract
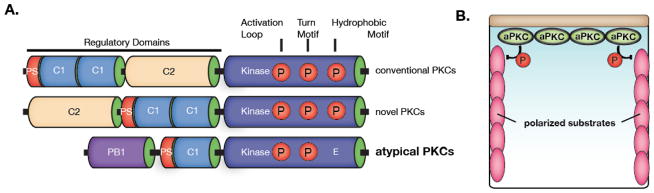
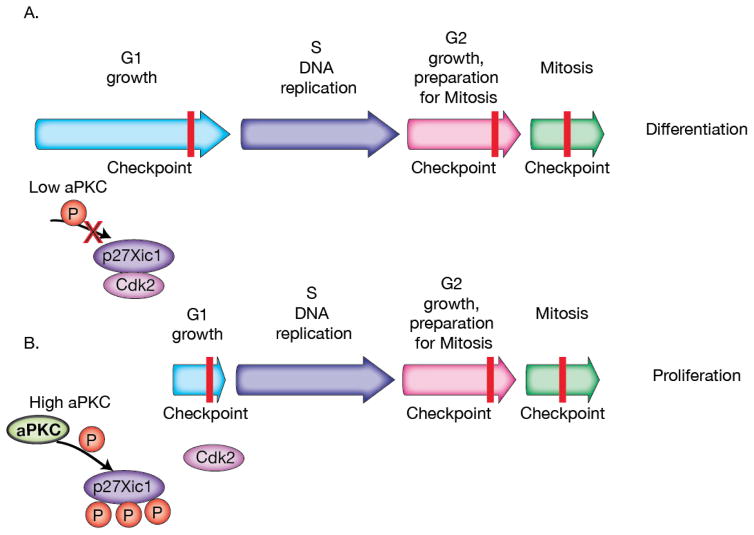
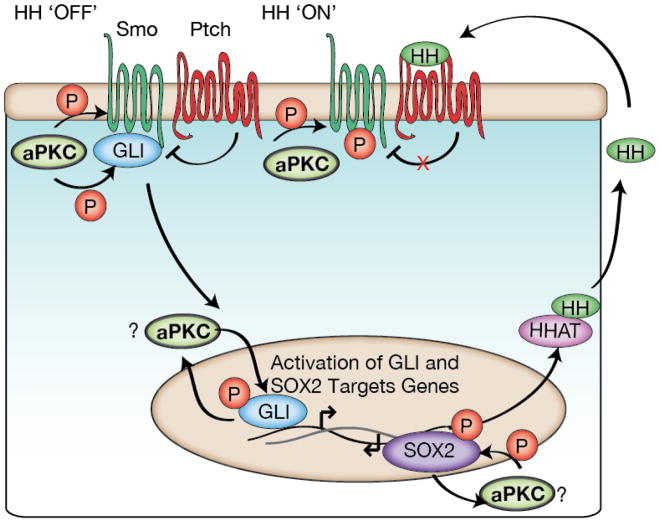
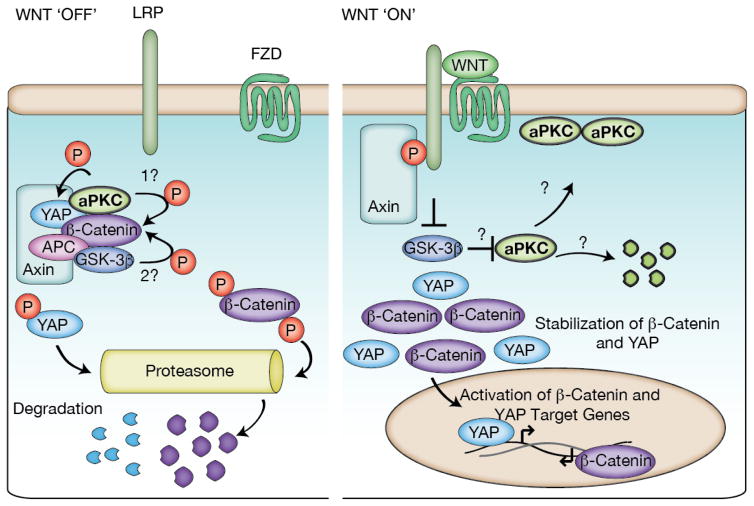
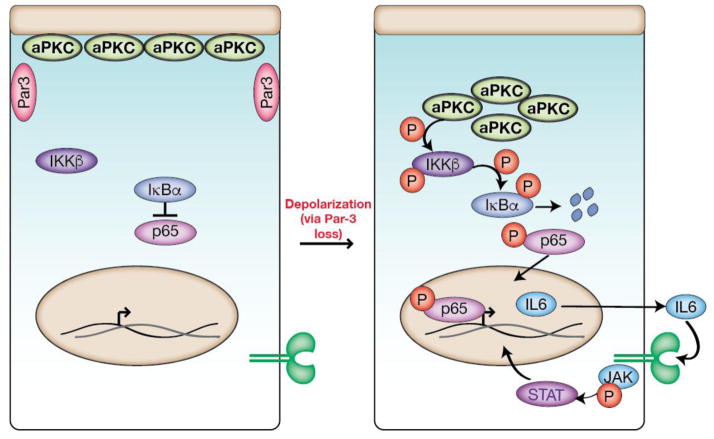
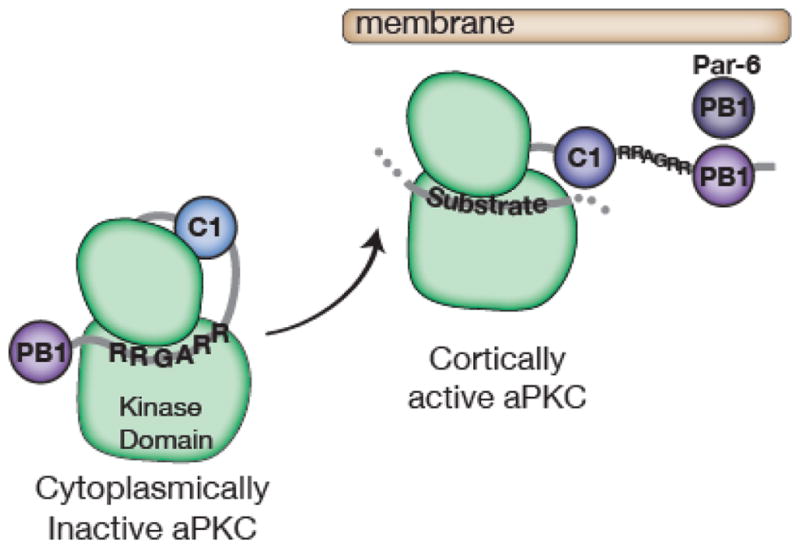
Complex organisms are faced with the challenge of generating and maintaining diverse cell types, ranging from simple epithelia to neurons and motile immune cells [1-3]. To meet this challenge, a complex set of regulatory pathways controls nearly every aspect of cell growth and function, including genetic and epigenetic programming, cytoskeleton dynamics, and protein trafficking. The far reach of cell fate specification pathways makes it particularly catastrophic when they malfunction, both during development and for tissue homeostasis in adult organisms. Furthermore, the therapeutic promise of stem cells derives from their ability to deftly navigate the multitude of pathways that control cell fate [4]. How the molecular components making up these pathways function to specify cell fate is beginning to become clear. Work from diverse systems suggests that the atypical Protein Kinase C (aPKC) is a key regulator of cell fate decisions in metazoans [5-7]. Here, we examine some of the diverse physiological outcomes of aPKC's function in differentiation, along with the molecular pathways that control aPKC and those that are responsive to changes in its catalytic activity.
Keywords: cell proliferation; differentiation; protein kinase.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
