

Characterization of multiple sequence alignment errors using complete-likelihood score and position-shift map
- PMID: 26992851
- PMCID: PMC4799563
- DOI: 10.1186/s12859-016-0945-5
Characterization of multiple sequence alignment errors using complete-likelihood score and position-shift map
Abstract
Background: Reconstruction of multiple sequence alignments (MSAs) is a crucial step in most homology-based sequence analyses, which constitute an integral part of computational biology. To improve the accuracy of this crucial step, it is essential to better characterize errors that state-of-the-art aligners typically make. For this purpose, we here introduce two tools: the complete-likelihood score and the position-shift map.
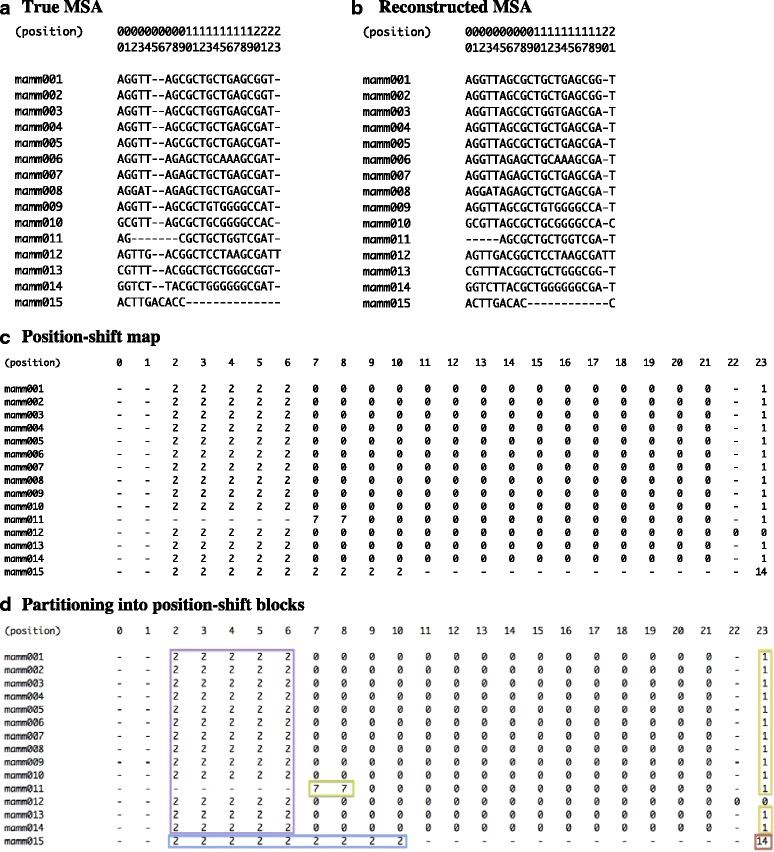
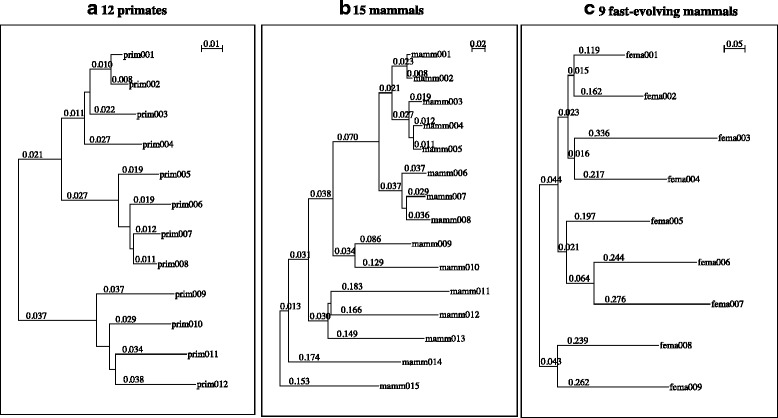
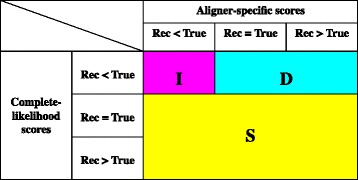
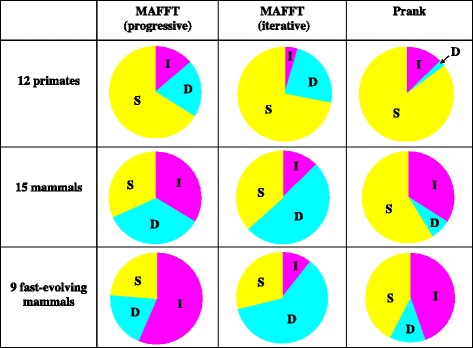
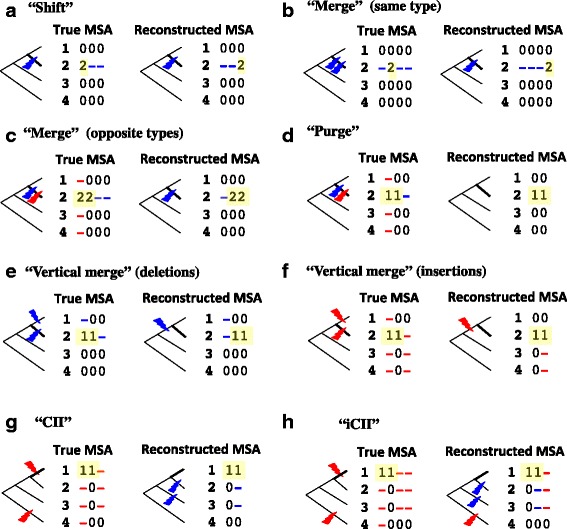
Results: The logarithm of the total probability of a MSA under a stochastic model of sequence evolution along a time axis via substitutions, insertions and deletions (called the "complete-likelihood score" here) can serve as an ideal score of the MSA. A position-shift map, which maps the difference in each residue's position between two MSAs onto one of them, can clearly visualize where and how MSA errors occurred and help disentangle composite errors. To characterize MSA errors using these tools, we constructed three sets of simulated MSAs of selectively neutral mammalian DNA sequences, with small, moderate and large divergences, under a stochastic evolutionary model with an empirically common power-law insertion/deletion length distribution. Then, we reconstructed MSAs using MAFFT and Prank as representative state-of-the-art single-optimum-search aligners. About 40-99% of the hundreds of thousands of gapped segments were involved in alignment errors. In a substantial fraction, from about 1/4 to over 3/4, of erroneously reconstructed segments, reconstructed MSAs by each aligner showed complete-likelihood scores not lower than those of the true MSAs. Out of the remaining errors, a majority by an iterative option of MAFFT showed discrepancies between the aligner-specific score and the complete-likelihood score, and a majority by Prank seemed due to inadequate exploration of the MSA space. Analyses by position-shift maps indicated that true MSAs are in considerable neighborhoods of reconstructed MSAs in about 80-99% of the erroneous segments for small and moderate divergences, but in only a minority for large divergences.
Conclusions: The results of this study suggest that measures to further improve the accuracy of reconstructed MSAs would substantially differ depending on the types of aligners. They also re-emphasize the importance of obtaining a probability distribution of fairly likely MSAs, instead of just searching for a single optimum MSA.
Keywords: Error; Insertion/deletion (indel); Likelihood; MSA space exploration; Multiple sequence alignment (MSA); Power-law; Probability distribution; Stochastic evolutionary model; Visualization.
Figures

References
-
- Felsenstein J. Inferring phylogenies. Sunderland (MA): Sinauer Associates; 2004.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
