

Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis
- PMID: 26993267
- PMCID: PMC4862068
- DOI: 10.1136/jmedgenet-2015-103263
Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis
Abstract
Background: We sought to investigate the diagnostic yield and mutation spectrum in previously reported genes for early-onset epilepsy and disorders of severe developmental delay.
Methods: In 400 patients with these disorders with no known underlying aetiology and no major structural brain anomaly, we analysed 46 genes using a combination of targeted sequencing on an Illumina MiSeq platform and targeted, exon-level microarray copy number analysis.
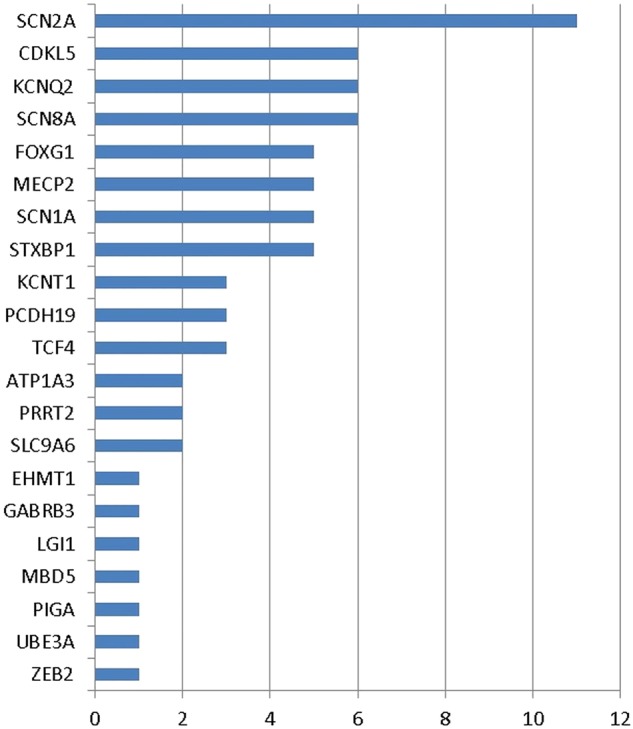
Results: We identified causative mutations in 71/400 patients (18%). The diagnostic rate was highest among those with seizure onset within the first two months of life (39%), although overall it was similar in those with and without seizures. The most frequently mutated gene was SCN2A (11 patients, 3%). Other recurrently mutated genes included CDKL5, KCNQ2, SCN8A (six patients each), FOXG1, MECP2, SCN1A, STXBP1 (five patients each), KCNT1, PCDH19, TCF4 (three patients each) and ATP1A3, PRRT2 and SLC9A6 (two patients each). Mutations in EHMT1, GABRB3, LGI1, MBD5, PIGA, UBE3A and ZEB2 were each found in single patients. We found mutations in a number of genes in patients where either the electroclinical features or dysmorphic phenotypes were atypical for the identified gene. In only 11 cases (15%) had the clinician sufficient certainty to specify the mutated gene as the likely cause before testing.
Conclusions: Our data demonstrate the considerable utility of a gene panel approach in the diagnosis of patients with early-onset epilepsy and severe developmental delay disorders., They provide further insights into the phenotypic spectrum and genotype-phenotype correlations for a number of the causative genes and emphasise the value of exon-level copy number testing in their analysis.
Keywords: Copy-number; Diagnostics; Epilepsy and seizures.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/
Figures
References
-
- Joint Epilepsy Council. 2011. http://www.jointepilepsycouncil.org.uk/
-
- Nava C, Dalle C, Rastetter A, Striano P, de Kovel CG, Nabbout R, Cancès C, Ville D, Brilstra EH, Gobbi G, Raffo E, Bouteiller D, Marie Y, Trouillard O, Robbiano A, Keren B, Agher D, Roze E, Lesage S, Nicolas A, Brice A, Baulac M, Vogt C, El Hajj N, Schneider E, Suls A, Weckhuysen S, Gormley P, Lehesjoki AE, De Jonghe P, Helbig I, Baulac S, Zara F, Koeleman BP; EuroEPINOMICS RES Consortium, Haaf T, LeGuern E, Depienne C. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet 2014;46:640–5. 10.1038/ng.2952 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials