

Pathogenic Variants in PIGG Cause Intellectual Disability with Seizures and Hypotonia
- PMID: 26996948
- PMCID: PMC4833197
- DOI: 10.1016/j.ajhg.2016.02.007
Pathogenic Variants in PIGG Cause Intellectual Disability with Seizures and Hypotonia
Abstract
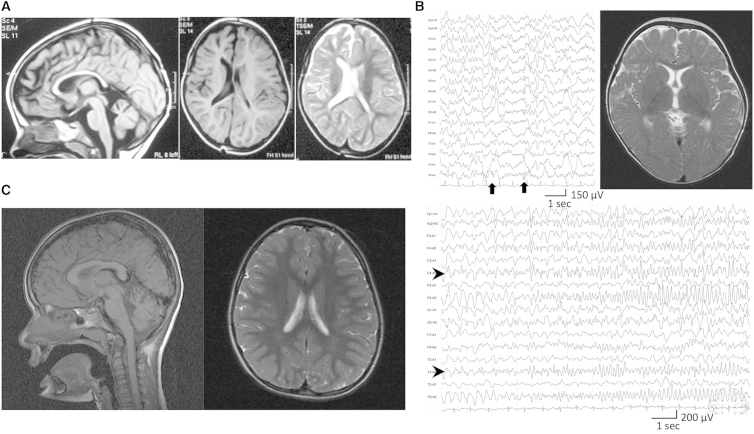
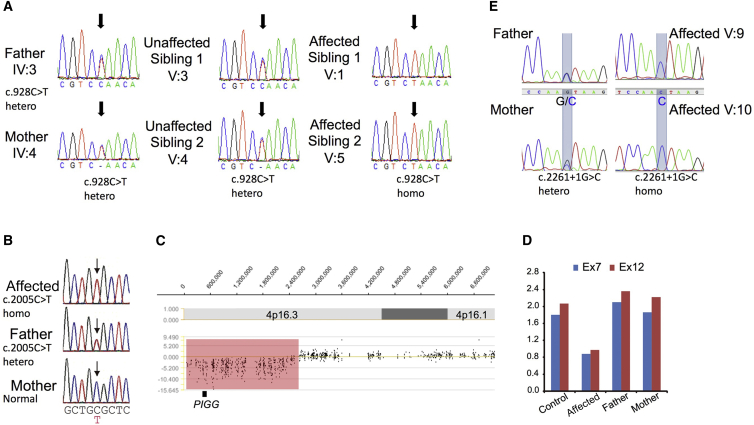
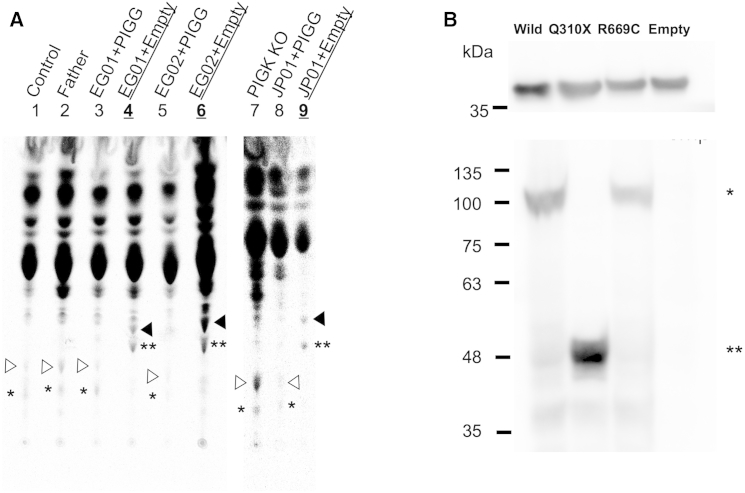
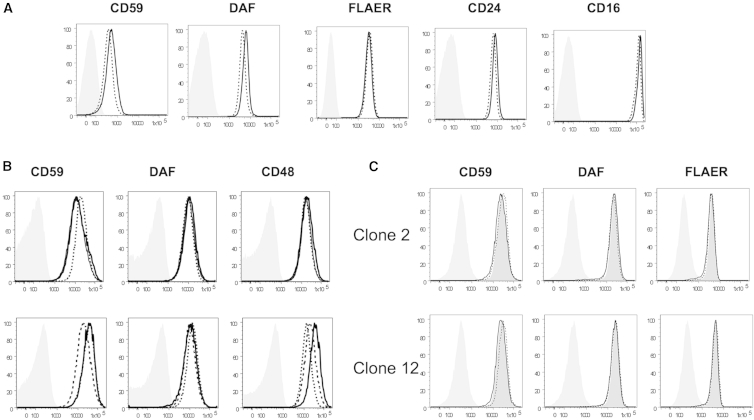
Glycosylphosphatidylinositol (GPI) is a glycolipid that anchors >150 various proteins to the cell surface. At least 27 genes are involved in biosynthesis and transport of GPI-anchored proteins (GPI-APs). To date, mutations in 13 of these genes are known to cause inherited GPI deficiencies (IGDs), and all are inherited as recessive traits. IGDs mainly manifest as intellectual disability, epilepsy, coarse facial features, and multiple organ anomalies. These symptoms are caused by the decreased surface expression of GPI-APs or by structural abnormalities of GPI. Here, we present five affected individuals (from two consanguineous families from Egypt and Pakistan and one non-consanguineous family from Japan) who show intellectual disability, hypotonia, and early-onset seizures. We identified pathogenic variants in PIGG, a gene in the GPI pathway. In the consanguineous families, homozygous variants c.928C>T (p.Gln310(∗)) and c.2261+1G>C were found, whereas the Japanese individual was compound heterozygous for c.2005C>T (p.Arg669Cys) and a 2.4 Mb deletion involving PIGG. PIGG is the enzyme that modifies the second mannose with ethanolamine phosphate, which is removed soon after GPI is attached to the protein. Physiological significance of this transient modification has been unclear. Using B lymphoblasts from affected individuals of the Egyptian and Japanese families, we revealed that PIGG activity was almost completely abolished; however, the GPI-APs had normal surface levels and normal structure, indicating that the pathogenesis of PIGG deficiency is not yet fully understood. The discovery of pathogenic variants in PIGG expands the spectrum of IGDs and further enhances our understanding of this etiopathogenic class of intellectual disability.
Copyright © 2016 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Maulik P.K., Mascarenhas M.N., Mathers C.D., Dua T., Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res. Dev. Disabil. 2011;32:419–436. - PubMed
-
- American Psychiatric Association (2013). DSM-5 Intellectual Disability Fact Sheet, http://www.dsm5.org/documents/intellectual%20disability%20fact%20sheet.pdf.
-
- World Health Organization Division of Mental Health and Prevention of Substance Abuse (1996). ICD-10 Guide for Mental Retardation, http://www.who.int/mental_health/media/en/69.pdf.
-
- Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
