

MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets
- PMID: 27004904
- PMCID: PMC8210823
- DOI: 10.1093/molbev/msw054
MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets
Abstract
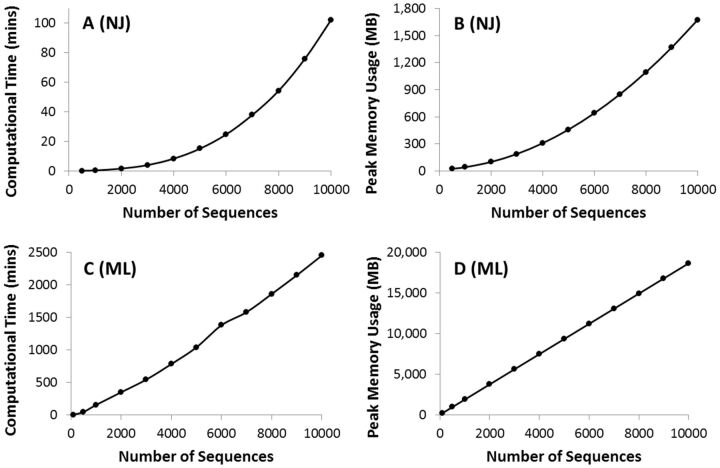
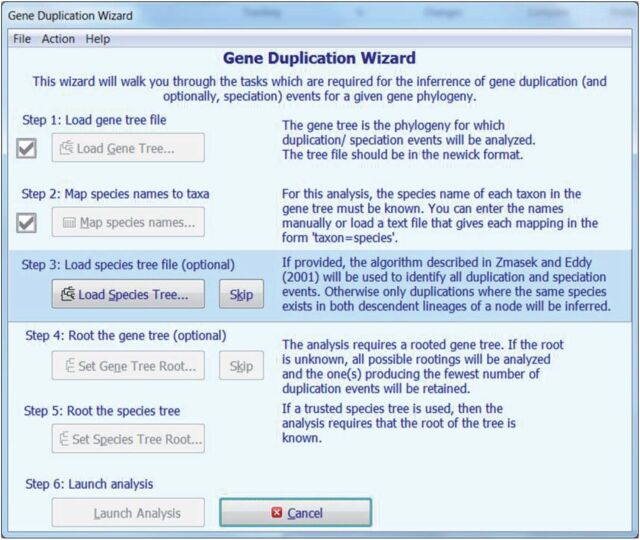
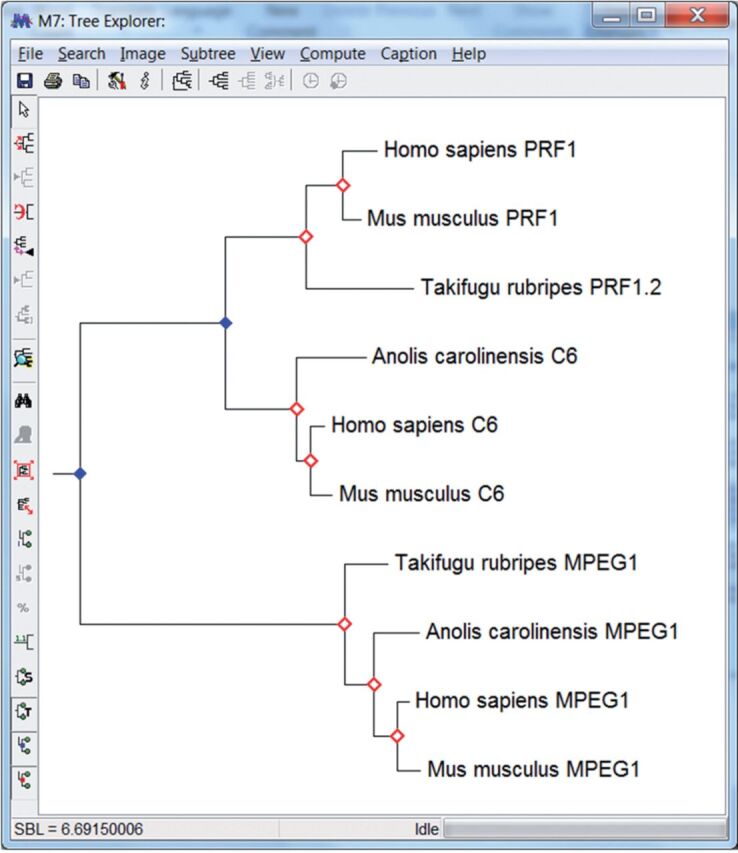
We present the latest version of the Molecular Evolutionary Genetics Analysis (Mega) software, which contains many sophisticated methods and tools for phylogenomics and phylomedicine. In this major upgrade, Mega has been optimized for use on 64-bit computing systems for analyzing larger datasets. Researchers can now explore and analyze tens of thousands of sequences in Mega The new version also provides an advanced wizard for building timetrees and includes a new functionality to automatically predict gene duplication events in gene family trees. The 64-bit Mega is made available in two interfaces: graphical and command line. The graphical user interface (GUI) is a native Microsoft Windows application that can also be used on Mac OS X. The command line Mega is available as native applications for Windows, Linux, and Mac OS X. They are intended for use in high-throughput and scripted analysis. Both versions are available from www.megasoftware.net free of charge.
Keywords: evolution.; gene families; software; timetree.
© The Author 2016. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
Comment in
-
MEGA Evolutionary Software Re-Engineered to Handle Today's Big Data Demands.Mol Biol Evol. 2016 Jul;33(7):1887. doi: 10.1093/molbev/msw074. Epub 2016 May 2. Mol Biol Evol. 2016. PMID: 27189553 No abstract available.
References
-
- Kumar S Tamura K Nei M. 1994. . M ega : molecular evolutionary genetics analysis software for microcomputers . Comput Appl Biosci . 10 : 189 – 191 . - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
