

Water electrolysis on La(1-x)Sr(x)CoO(3-δ) perovskite electrocatalysts
- PMID: 27006166
- PMCID: PMC4814573
- DOI: 10.1038/ncomms11053
Water electrolysis on La(1-x)Sr(x)CoO(3-δ) perovskite electrocatalysts
Abstract
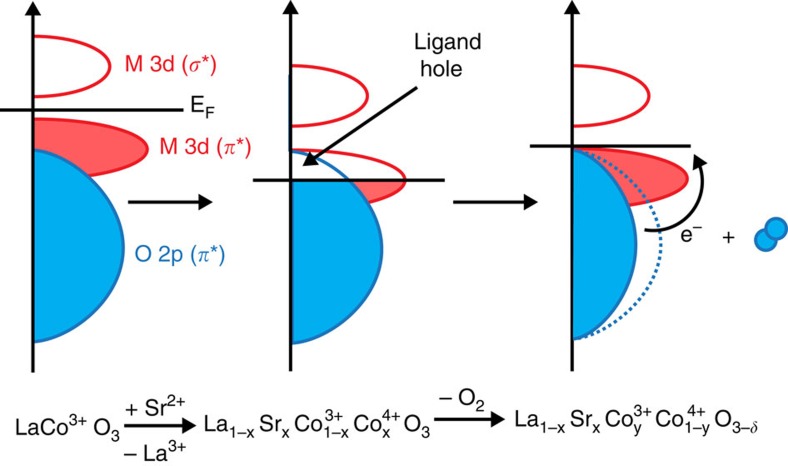
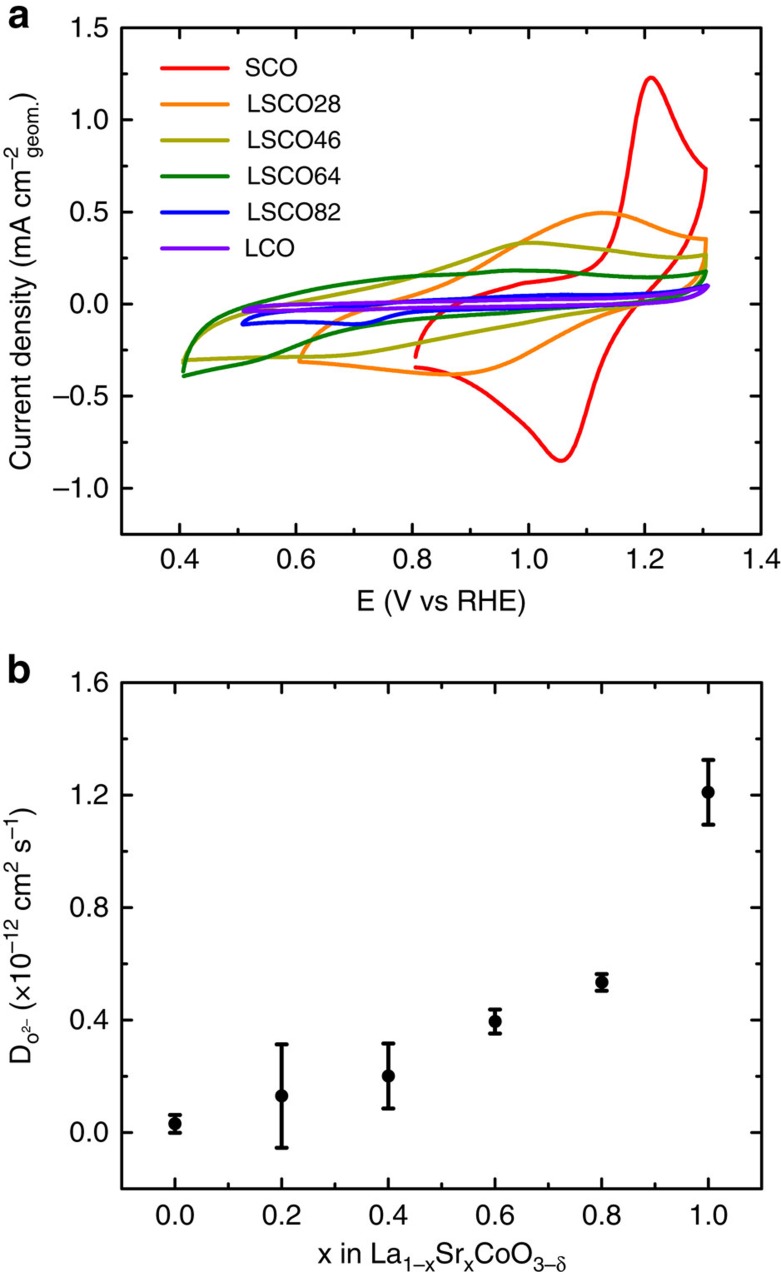
Perovskite oxides are attractive candidates as catalysts for the electrolysis of water in alkaline energy storage and conversion systems. However, the rational design of active catalysts has been hampered by the lack of understanding of the mechanism of water electrolysis on perovskite surfaces. Key parameters that have been overlooked include the role of oxygen vacancies, B-O bond covalency, and redox activity of lattice oxygen species. Here we present a series of cobaltite perovskites where the covalency of the Co-O bond and the concentration of oxygen vacancies are controlled through Sr(2+) substitution into La(1-x)Sr(x)CoO(3-δ) . We attempt to rationalize the high activities of La(1-x)Sr(x)CoO(3-δ) through the electronic structure and participation of lattice oxygen in the mechanism of water electrolysis as revealed through ab initio modelling. Using this approach, we report a material, SrCoO2.7, with a high, room temperature-specific activity and mass activity towards alkaline water electrolysis.
Figures
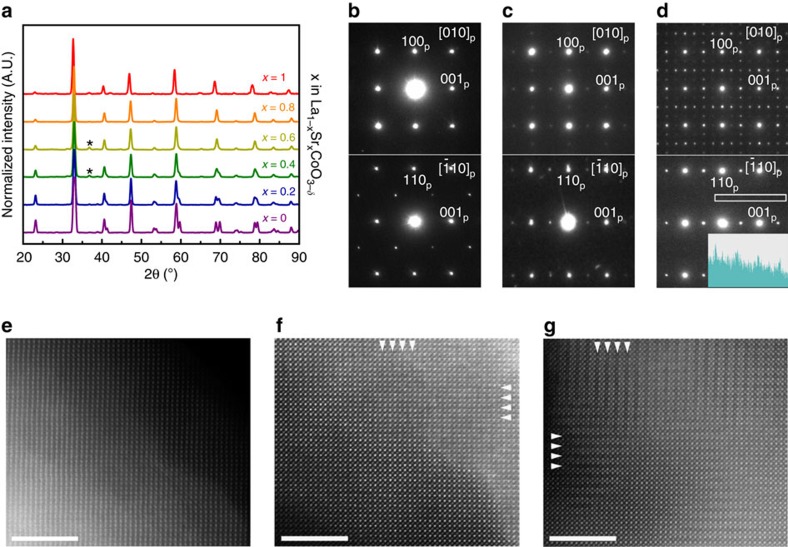
 SAED patterns of SCO, but the intensity profile (shown as insert in d) along the area marked with the white rectangle demonstrates their presence undoubtedly. (e–g) [010]p HAADF-STEM images of LSCO82 (e), LSCO28 (f) and SCO (g). The image of LSCO82 shows uniform perovskite structure, whereas the images of LSCO28 and SCO show faint darker stripes spaced by 2ap (marked by arrowheads) indicating nanoscale-twinned arrangement of the alternating (CoO2) perovskite layers and (CoO2−δ) anion-deficient layers. Scale bars are 5 nm.
SAED patterns of SCO, but the intensity profile (shown as insert in d) along the area marked with the white rectangle demonstrates their presence undoubtedly. (e–g) [010]p HAADF-STEM images of LSCO82 (e), LSCO28 (f) and SCO (g). The image of LSCO82 shows uniform perovskite structure, whereas the images of LSCO28 and SCO show faint darker stripes spaced by 2ap (marked by arrowheads) indicating nanoscale-twinned arrangement of the alternating (CoO2) perovskite layers and (CoO2−δ) anion-deficient layers. Scale bars are 5 nm.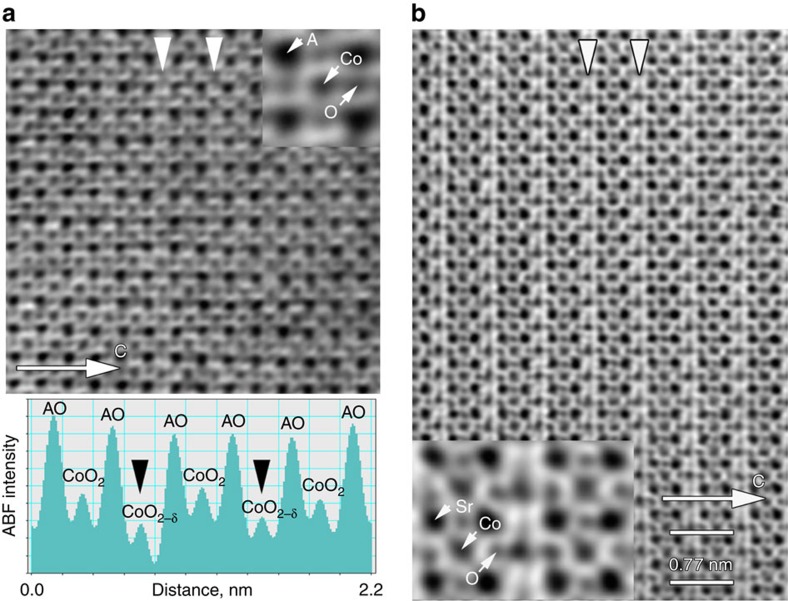
References
-
- Cui C., Gan L., Heggen M., Rudi S. & Strasser P. Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis. Nat. Mater. 12, 765–771 (2013). - PubMed
-
- Gupta G. et al. Highly stable and active Pt−Cu oxygen reduction electrocatalysts based on mesoporous graphitic carbon supports. Chem. Mater. 21, 4515–4526 (2009).
-
- James Patrick H. The Electrochemistry of Oxygen Interscience Publishers (1968).
-
- McCrory C. C. L., Jung S., Peters J. C. & Jaramillo T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 135, 16977–16987 (2013). - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
