

Patients with mosaic methylation patterns of the Prader-Willi/Angelman Syndrome critical region exhibit AS-like phenotypes with some PWS features
- PMID: 27006693
- PMCID: PMC4802915
- DOI: 10.1186/s13039-016-0233-0
Patients with mosaic methylation patterns of the Prader-Willi/Angelman Syndrome critical region exhibit AS-like phenotypes with some PWS features
Abstract
Background: Loss of expression of imprinted genes in the 15q11.2-q13 region is known to cause either Prader-Willi syndrome (PWS) or Angelman syndrome (AS), depending on the parent of origin. In some patients (1 % in PWS and 2-4 % in AS), the disease is due to aberrant imprinting or gene silencing, or both.
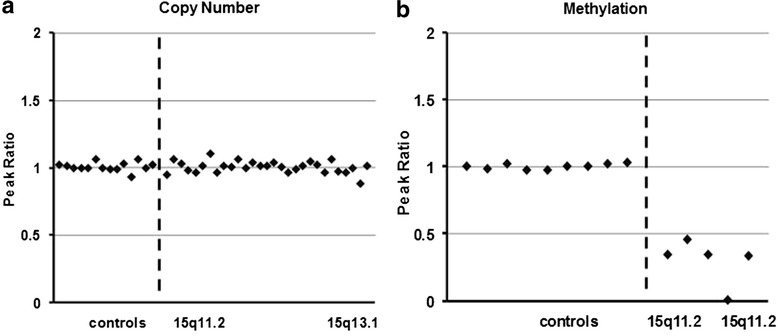
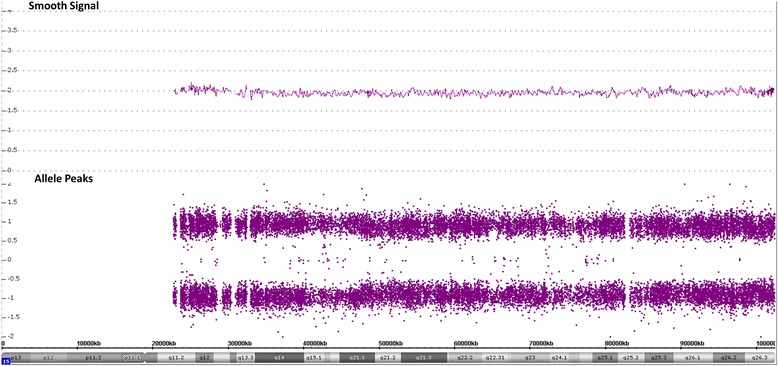
Results: We report here a 4-year-old boy on whom a chromosomal microarray (CMA) was performed due to mild hand tremors, mild developmental delays, and clumsiness. CMA revealed absence of heterozygosity (AOH) spanning the entire chromosome 15, suggesting uniparental isodisomy 15. The patient had no definitive phenotypic features of PWS or AS. Methylation-sensitive multiplex ligation-dependent probe amplification (MS-MLPA) was performed to determine the parent of origin of the uniparental disomy (UPD) by examining methylation status at maternally imprinted sites. Interestingly, our patient had a mosaic methylation pattern. We identified nine additional previously tested patients with a similar mosaic methylation pattern. CMA was performed on these individuals retrospectively to test whether patients with mosaic methylation are more likely to have UPD of chromosome 15. Of the nine patients, only one had regions of AOH on chromosome 15; however, this patient had numerous regions of AOH on multiple chromosomes suggestive of consanguinity.
Conclusion: The patients with mosaic methylation had milder or atypical features of AS, and the majority also had some features characteristic of PWS. We suggest that quantitative methylation analysis be performed for cases of atypical PWS or AS. It is also important to follow up with methylation testing when whole-chromosome isodisomy is detected.
Keywords: 15q11.2-q13; Chromosomal microarray; MS-MLPA; Mosaic methylation; PWASCR.
Figures
References
-
- Buiting K, Dittrich B, Gross S, Lich C, Farber C, Buchholz T, et al. Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet. 1998;63(1):170–80. doi: 10.1086/301935. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
