

Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21)
- PMID: 27008867
- PMCID: PMC5081059
- DOI: 10.1093/hmg/ddw096
Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21)
Abstract
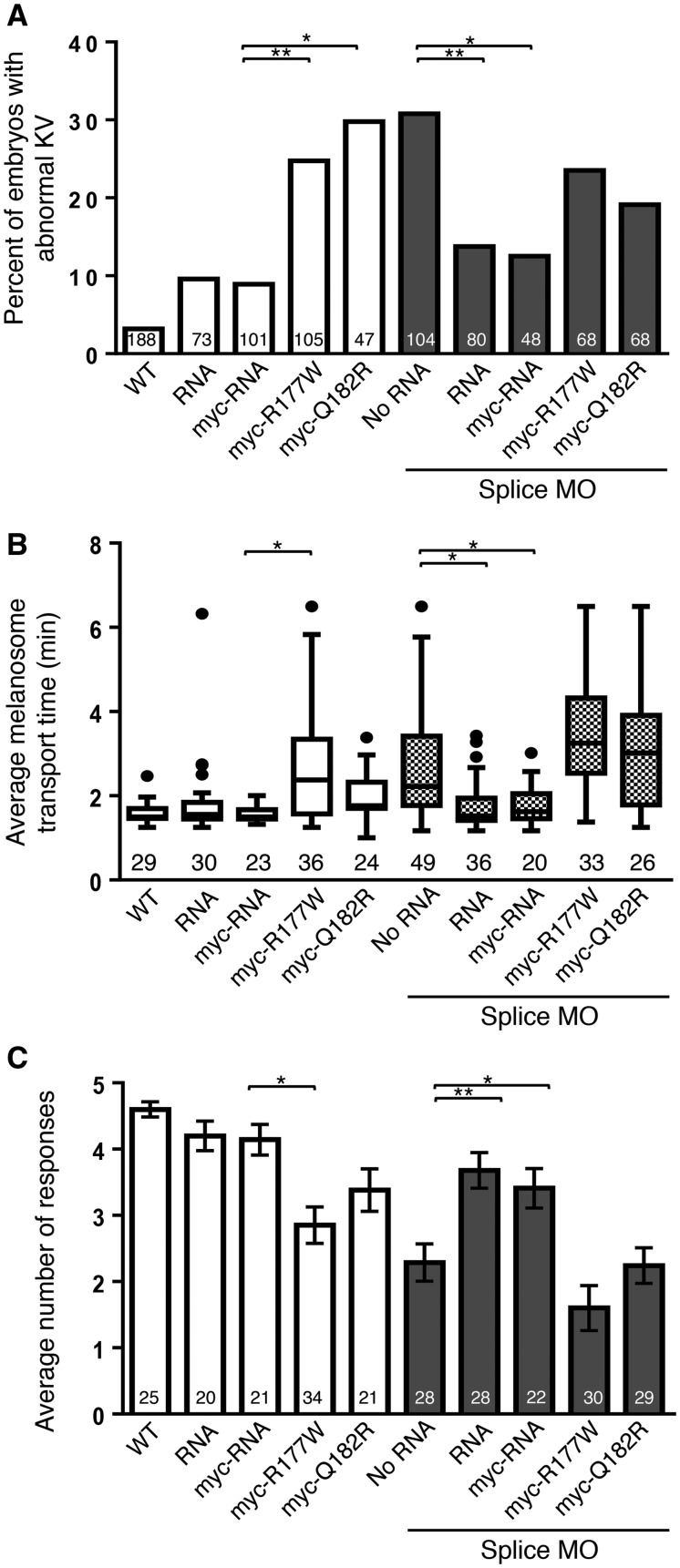
Bardet Biedl syndrome (BBS) is a multisystem genetically heterogeneous ciliopathy that most commonly leads to obesity, photoreceptor degeneration, digit anomalies, genito-urinary abnormalities, as well as cognitive impairment with autism, among other features. Sequencing of a DNA sample from a 17-year-old female affected with BBS did not identify any mutation in the known BBS genes. Whole-genome sequencing identified a novel loss-of-function disease-causing homozygous mutation (K102*) in C8ORF37, a gene coding for a cilia protein. The proband was overweight (body mass index 29.1) with a slowly progressive rod-cone dystrophy, a mild learning difficulty, high myopia, three limb post-axial polydactyly, horseshoe kidney, abnormally positioned uterus and elevated liver enzymes. Mutations in C8ORF37 were previously associated with severe autosomal recessive retinal dystrophies (retinitis pigmentosa RP64 and cone-rod dystrophy CORD16) but not BBS. To elucidate the functional role of C8ORF37 in a vertebrate system, we performed gene knockdown in Danio rerio and assessed the cardinal features of BBS and visual function. Knockdown of c8orf37 resulted in impaired visual behavior and BBS-related phenotypes, specifically, defects in the formation of Kupffer's vesicle and delays in retrograde transport. Specificity of these phenotypes to BBS knockdown was shown with rescue experiments. Over-expression of human missense mutations in zebrafish also resulted in impaired visual behavior and BBS-related phenotypes. This is the first functional validation and association of C8ORF37 mutations with the BBS phenotype, which identifies BBS21. The zebrafish studies hereby show that C8ORF37 variants underlie clinically diagnosed BBS-related phenotypes as well as isolated retinal degeneration.
© The Author 2016. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures





References
-
- Kerr E.N., Bhan A, Héon E. (2016) Exploration of the cognitive, adaptive and behavioral functioning of patients affected with Bardet-Biedl syndrome. Clin. Genet., 89, 426–433. - PubMed
-
- Kwitek-Black A.E., Carmi R., Duyk G.M., Buetow K.H., Elbedour K., Parvari R., Yandava C.N., Stone E.M., Sheffield V.C. (1993) Linkage of Bardet-Biedl syndrome to chromosome 16q and evidence for non-allelic genetic heterogeneity. Nat. Genet., 3, 392–396. - PubMed
-
- Leppert M., Baird L., Anderson K.L., Otterud B., Lupski J.R., Lewis R.A. (1994) Bardet-Biedl syndrome is linked to DNA markers on chromosome 11q and is genetically heterogeneous. Nat. Genet., 7, 108–112. - PubMed
-
- Sheffield V.C., Carmi R., Kwitek-Black A., Rokhlina T., Nishimura D., Duyk G.M., Elbedour K., Sunden S.L., Stone E.M. (1994) Identification of a Bardet-Biedl syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mapping. Hum. Mol. Genet., 3, 1331–1335. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
