

Genetically Diverse Low Pathogenicity Avian Influenza A Virus Subtypes Co-Circulate among Poultry in Bangladesh
- PMID: 27010791
- PMCID: PMC4806916
- DOI: 10.1371/journal.pone.0152131
Genetically Diverse Low Pathogenicity Avian Influenza A Virus Subtypes Co-Circulate among Poultry in Bangladesh
Abstract
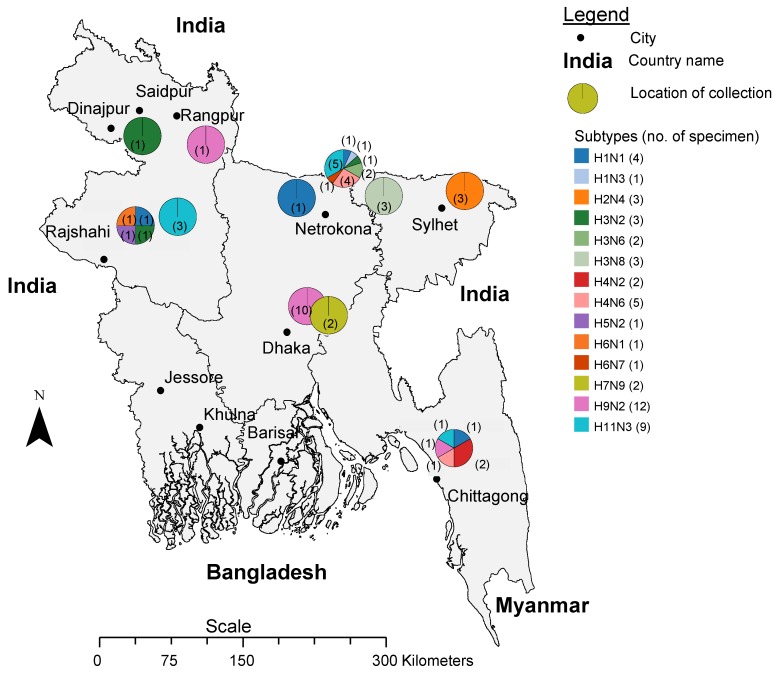
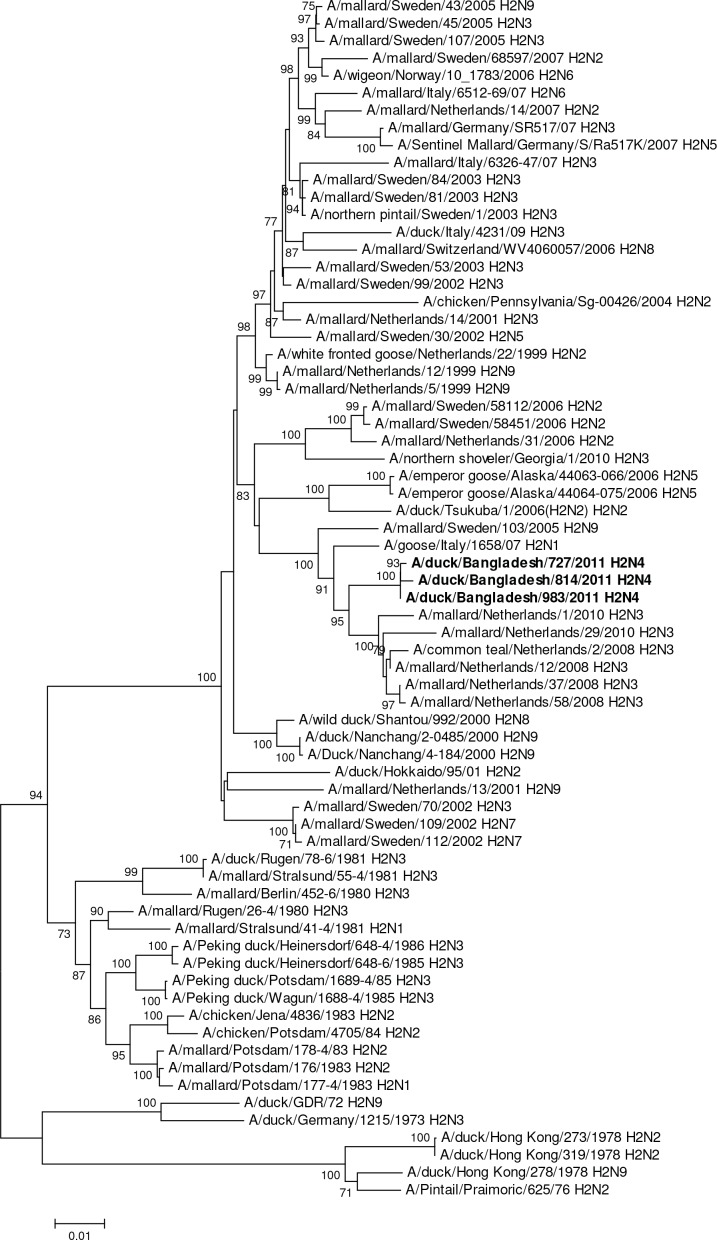
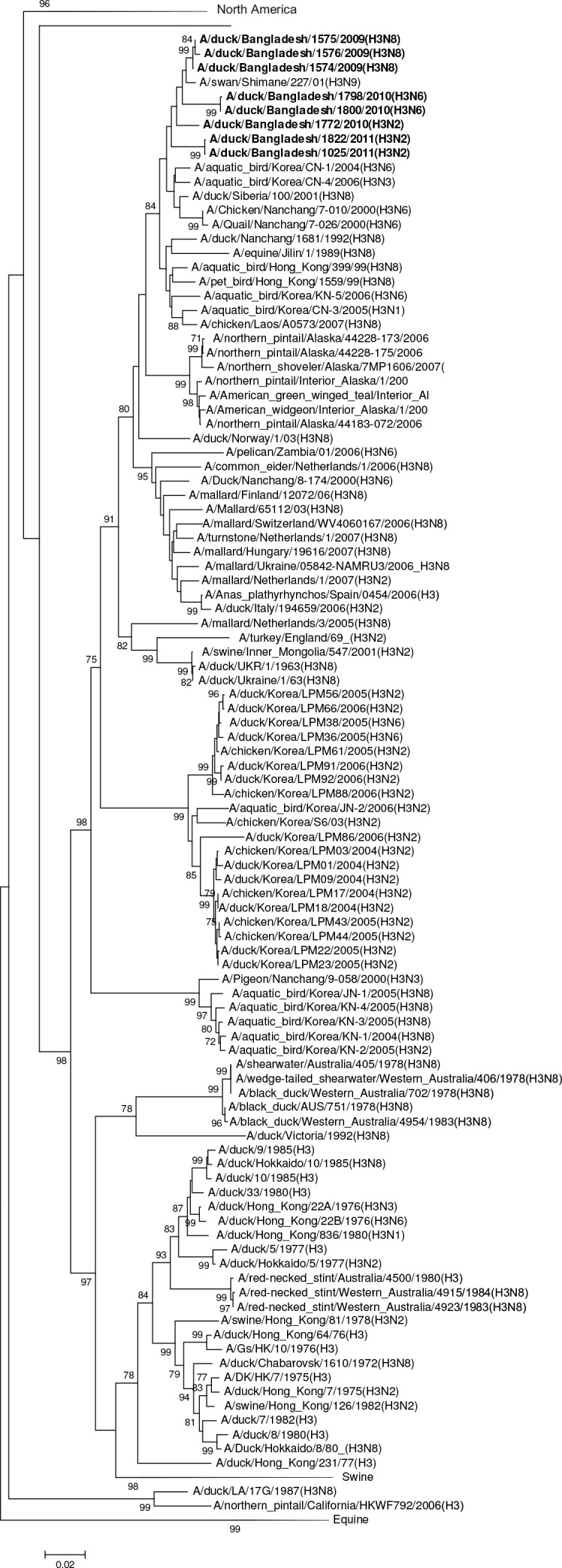
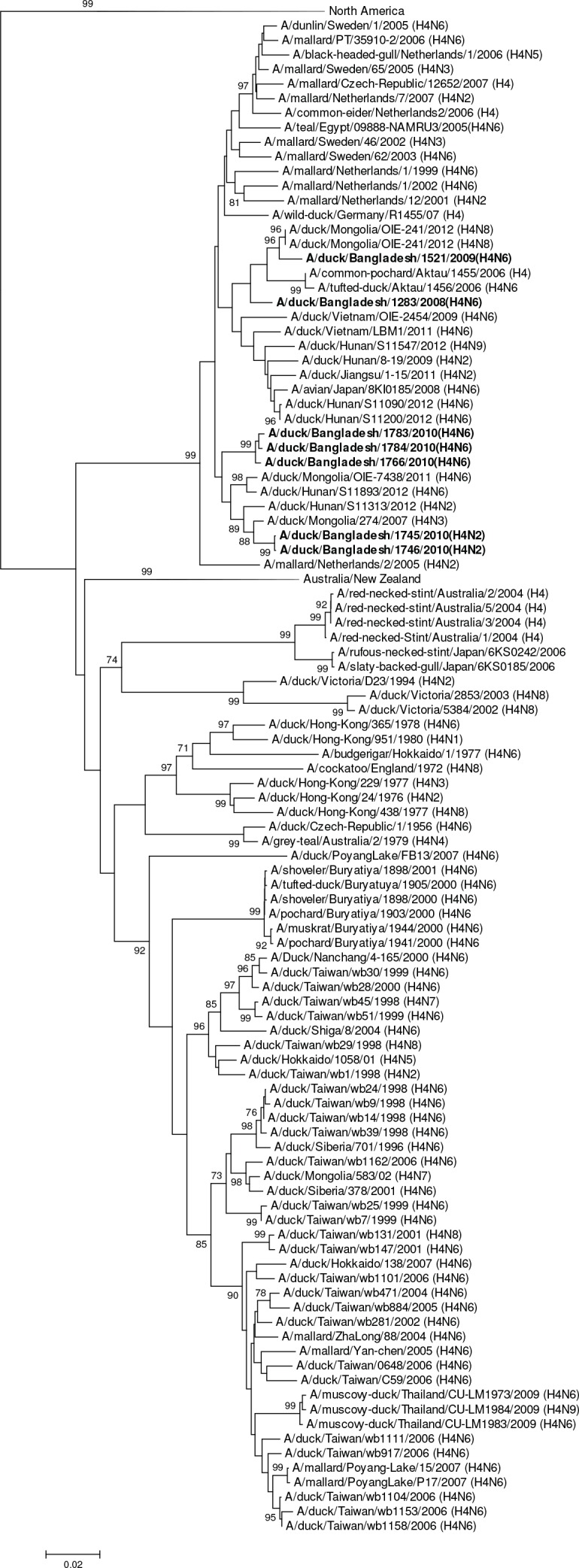
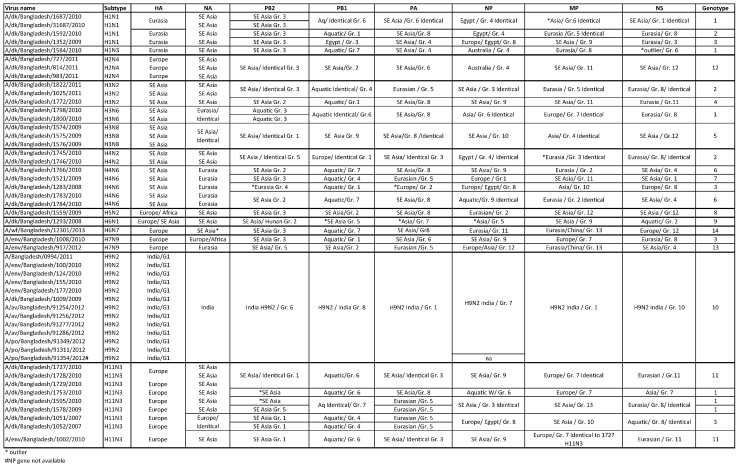
Influenza virus surveillance, poultry outbreak investigations and genomic sequencing were assessed to understand the ecology and evolution of low pathogenicity avian influenza (LPAI) A viruses in Bangladesh from 2007 to 2013. We analyzed 506 avian specimens collected from poultry in live bird markets and backyard flocks to identify influenza A viruses. Virus isolation-positive specimens (n = 50) were subtyped and their coding-complete genomes were sequenced. The most frequently identified subtypes among LPAI isolates were H9N2, H11N3, H4N6, and H1N1. Less frequently detected subtypes included H1N3, H2N4, H3N2, H3N6, H3N8, H4N2, H5N2, H6N1, H6N7, and H7N9. Gene sequences were compared to publicly available sequences using phylogenetic inference approaches. Among the 14 subtypes identified, the majority of viral gene segments were most closely related to poultry or wild bird viruses commonly found in Southeast Asia, Europe, and/or northern Africa. LPAI subtypes were distributed over several geographic locations in Bangladesh, and surface and internal protein gene segments clustered phylogenetically with a diverse number of viral subtypes suggesting extensive reassortment among these LPAI viruses. H9N2 subtype viruses differed from other LPAI subtypes because genes from these viruses consistently clustered together, indicating this subtype is enzootic in Bangladesh. The H9N2 strains identified in Bangladesh were phylogenetically and antigenically related to previous human-derived H9N2 viruses detected in Bangladesh representing a potential source for human infection. In contrast, the circulating LPAI H5N2 and H7N9 viruses were both phylogenetically and antigenically unrelated to H5 viruses identified previously in humans in Bangladesh and H7N9 strains isolated from humans in China. In Bangladesh, domestic poultry sold in live bird markets carried a wide range of LPAI virus subtypes and a high diversity of genotypes. These findings, combined with the seven year timeframe of sampling, indicate a continuous circulation of these viruses in the country.
Conflict of interest statement
Figures

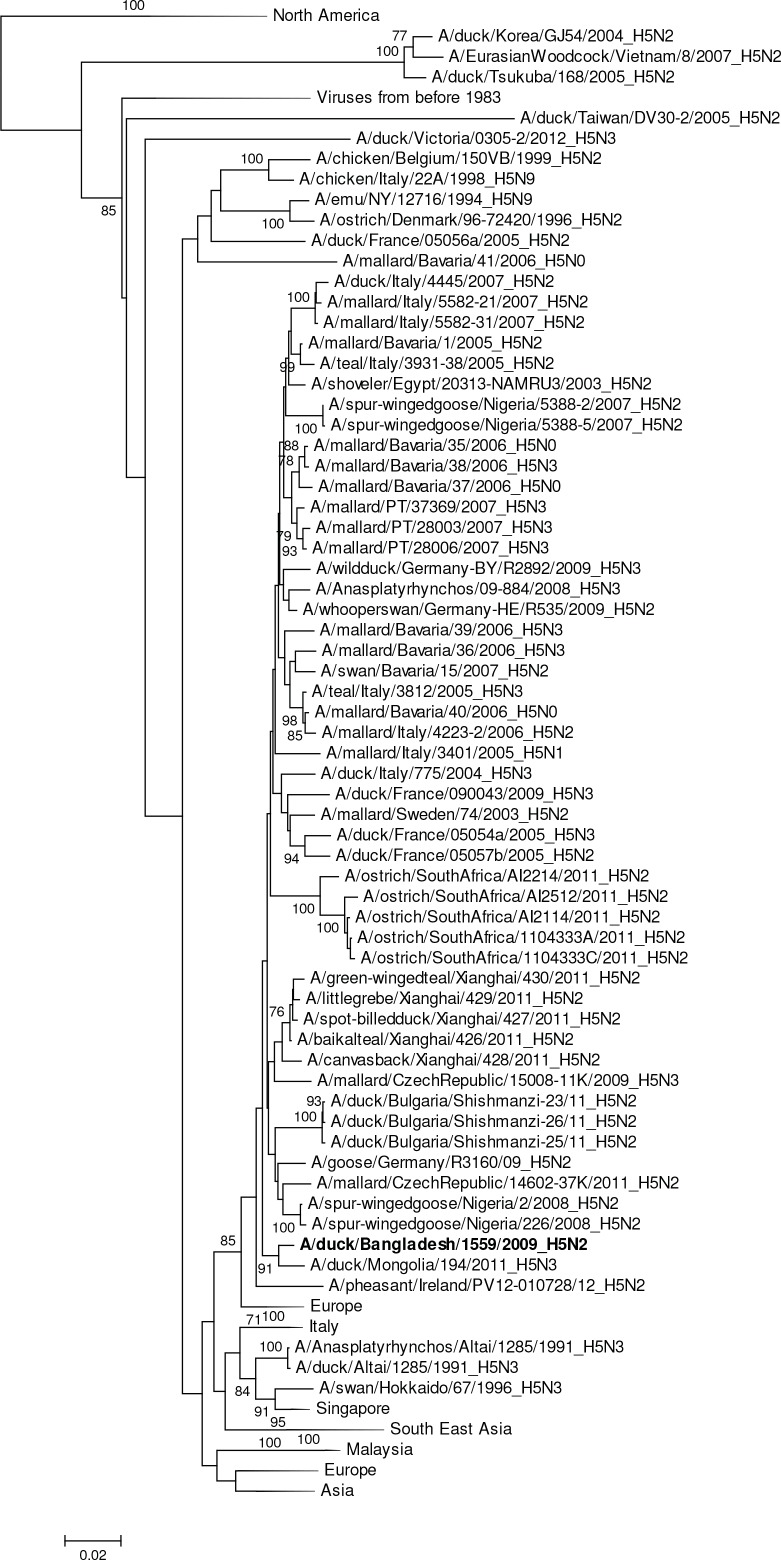
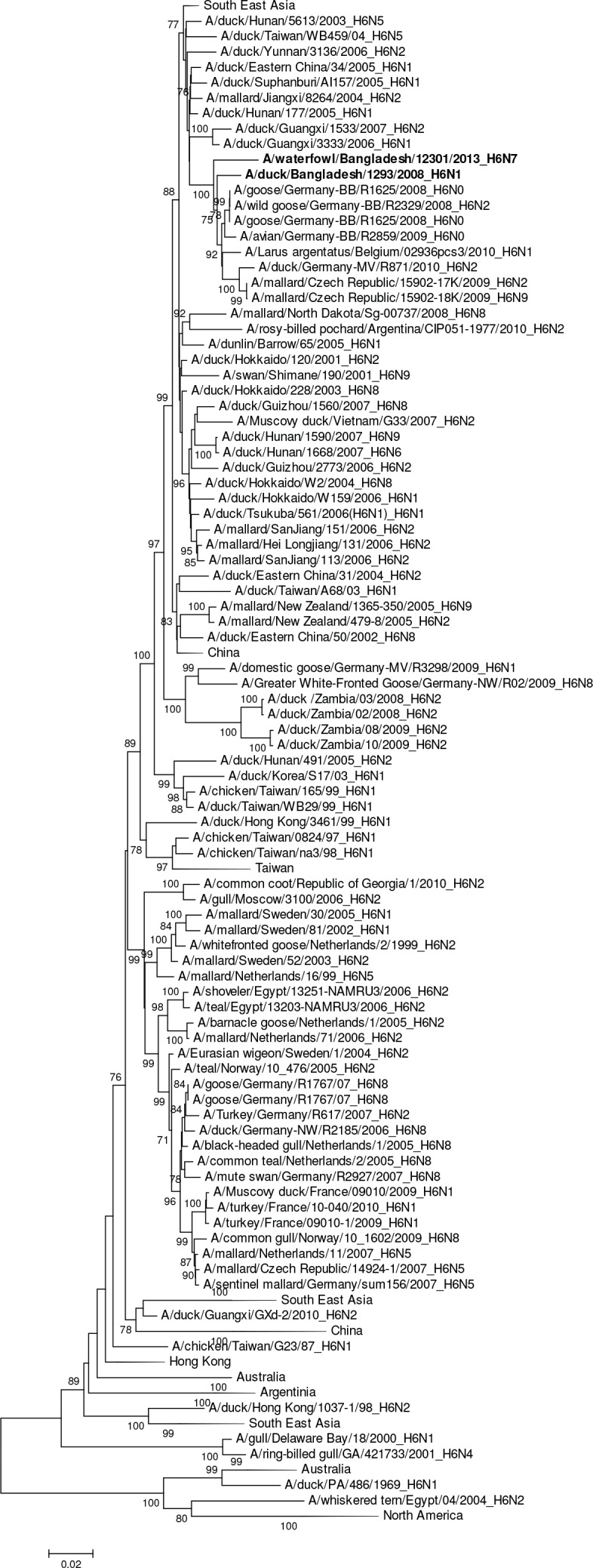
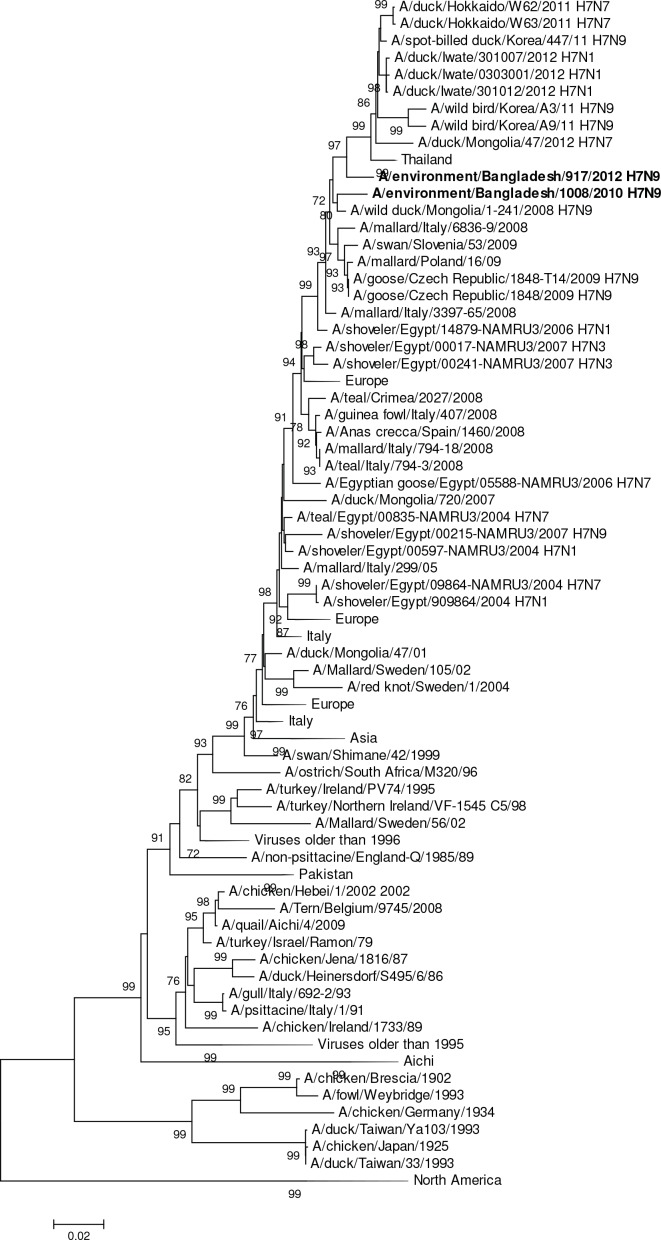
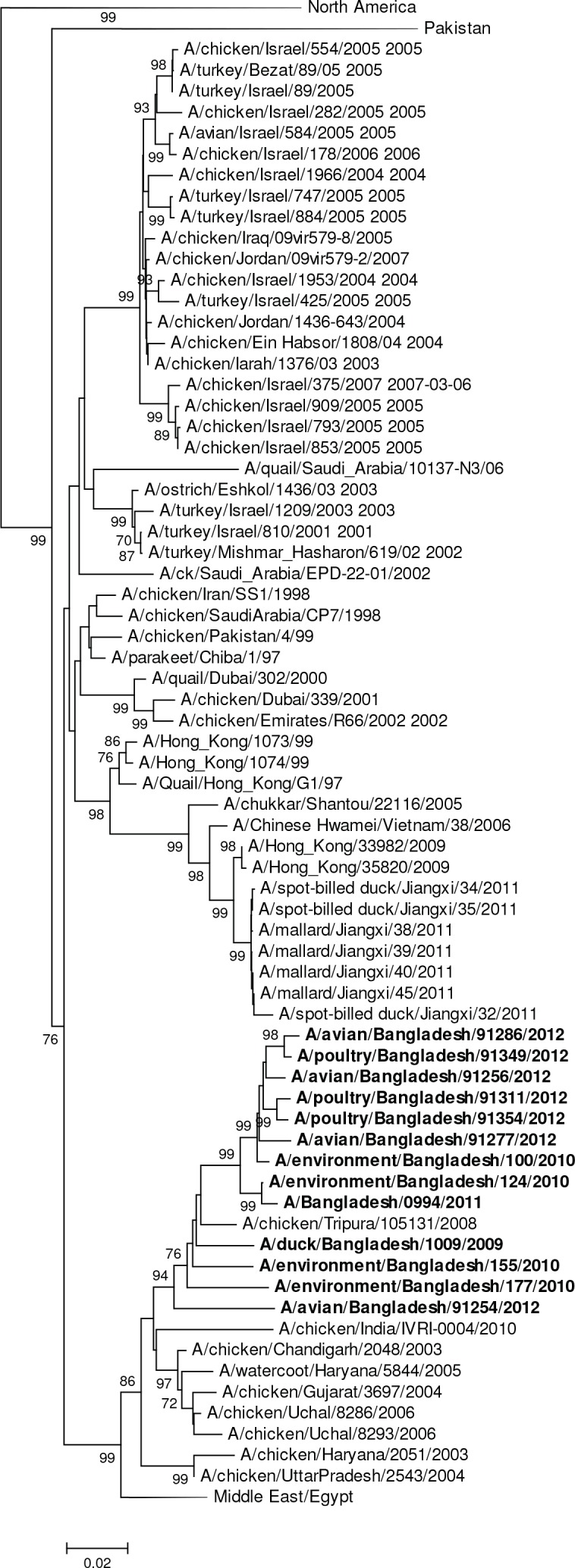

References
-
- Alexander DJ. An overview of the epidemiology of avian influenza. Vaccine. 2007. July 26;25(30):5637–44. . Epub 2006/11/28. eng. - PubMed
-
- OIE. Highly pathogenic avian influenza country information and timelines 2013 [cited 2015 May 20]. Available from: http://www.oie.int/wahis_2/public/wahid.php/Countryinformation/Countryti....
-
- OIE. AVIAN INFLUENZA Terrestrial Manual [Internet]. 2014 June 24 [cited 2015 June 24]; (CHAPTER 2.3.4.). Available from: http://www.oie.int/fileadmin/Home/fr/Health_standards/tahm/2.03.04_AI.pdf.
-
- WHO/GIP. Cumulative number of confirmed human cases for avian influenza A(H5N1) reported to WHO, 2003–2015 Geneva: WHO, 2015. 31 March. Report No.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
