

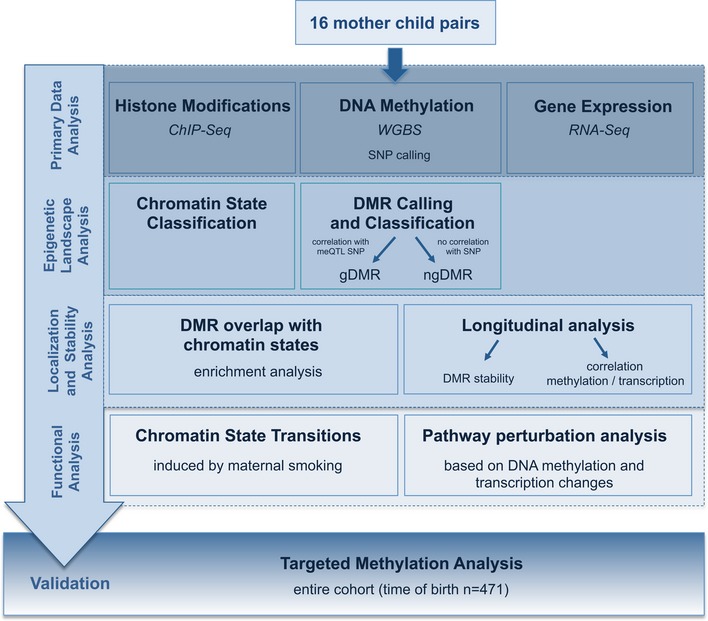
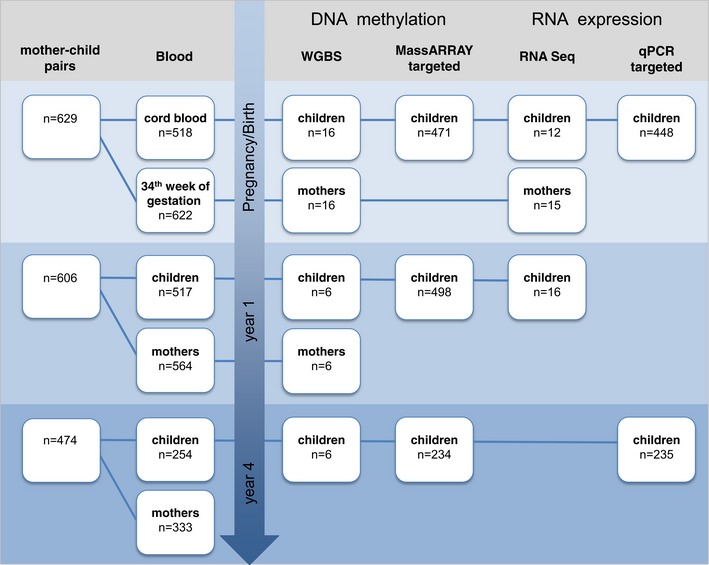
Environment-induced epigenetic reprogramming in genomic regulatory elements in smoking mothers and their children
- PMID: 27013061
- PMCID: PMC4812527
- DOI: 10.15252/msb.20156520
Environment-induced epigenetic reprogramming in genomic regulatory elements in smoking mothers and their children
Abstract
Epigenetic mechanisms have emerged as links between prenatal environmental exposure and disease risk later in life. Here, we studied epigenetic changes associated with maternal smoking at base pair resolution by mapping DNA methylation, histone modifications, and transcription in expectant mothers and their newborn children. We found extensive global differential methylation and carefully evaluated these changes to separate environment associated from genotype-related DNA methylation changes. Differential methylation is enriched in enhancer elements and targets in particular "commuting" enhancers having multiple, regulatory interactions with distal genes. Longitudinal whole-genome bisulfite sequencing revealed that DNA methylation changes associated with maternal smoking persist over years of life. Particularly in children prenatal environmental exposure leads to chromatin transitions into a hyperactive state. Combined DNA methylation, histone modification, and gene expression analyses indicate that differential methylation in enhancer regions is more often functionally translated than methylation changes in promoters or non-regulatory elements. Finally, we show that epigenetic deregulation of a commuting enhancer targeting c-Jun N-terminal kinase 2 (JNK2) is linked to impaired lung function in early childhood.
Keywords: WGBS; enhancer deregulation; environment; epigenetics; histone modifications.
© 2016 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
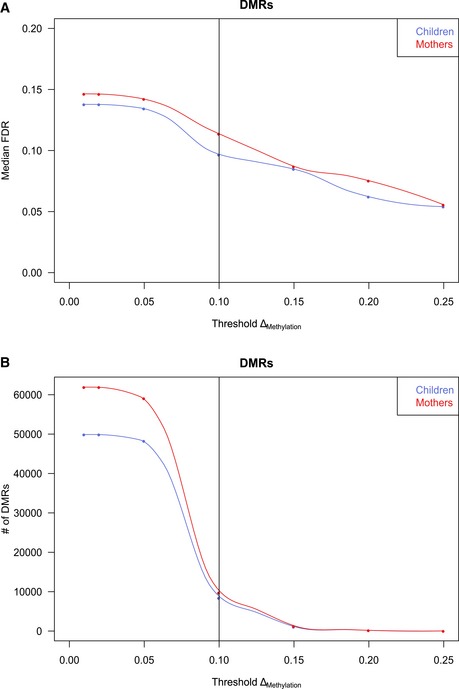
Median false discovery rate (FDR) estimated by permutation analysis at different cutoffs for mean methylation changes of DMRs (ΔMethylation).
Number of DMRs after filtering called at different mean methylation changes. The 10% ΔMethylation cutoff is marked and indicates a fair balance between sensitivity and specificity while offering acceptable FDRs (9.7% in children, 11.4% in mothers).
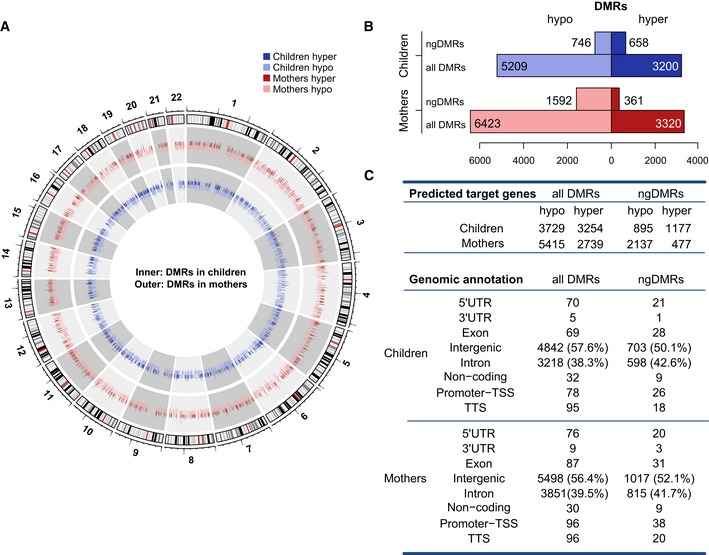
Circular representation of DNA methylation levels for mothers (outer circle) and children (inner circle). The height of each bar indicates the methylation change between the smoking and non‐smoking group (dark hue: hypermethylation, light hue: hypomethylation).
Bar plots represent the number of hypo‐ versus hypermethylated DMRs for all DMRs and ngDMRs in children and mothers.
Number of genes predicted to interact with DMRs and ngDMRs. Annotation of allDMRs/ngDMRs according to genomic categories (lower panel).
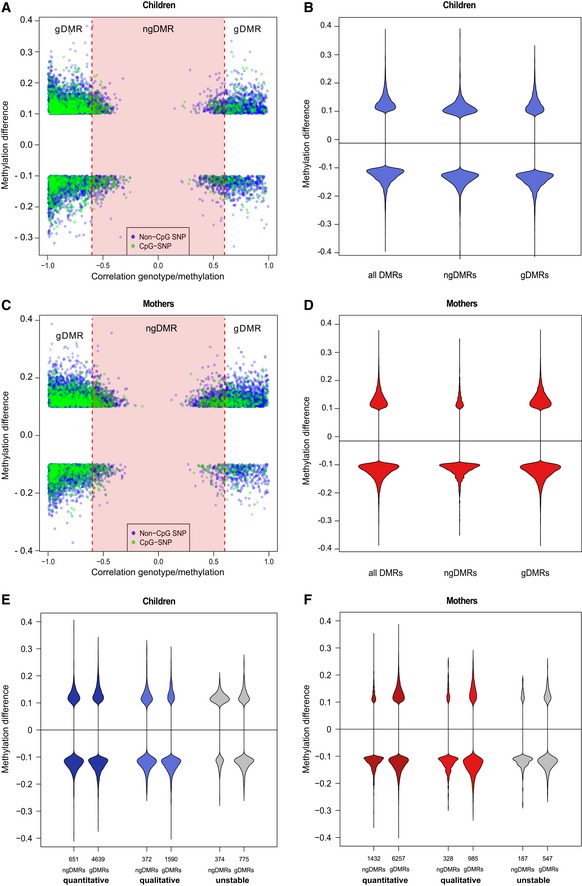
- A–D
Each DMR is associated with its most highly associated SNP within ± 5 kb, and each dot represents a DMR/SNP association. The x‐axis shows the level of correlation between genotype and methylation level, and the y‐axis shows the methylation difference for the DMR between the smoking and non‐smoking group. DMRs associated with a CpG‐destroying SNP are highlighted in green. A gDMR is defined as a DMR, which has an associated neighboring SNP with an absolute correlation above 0.6 (red vertical lines, FDR 10%), and the remaining DMRs are defined as ngDMRs (red shaded area). The effect of the 10% cutoff on the methylation difference is seen in this plot. Violin plots in (B, D) show the methylation difference in all DMRs, ngDMRs, and gDMRs.
- E, F
Methylation differences are shown for the various DMR stability classes, distinguishing ngDMRs (left in violin plot pairs) from gDMRs (right violin plot in each pair).
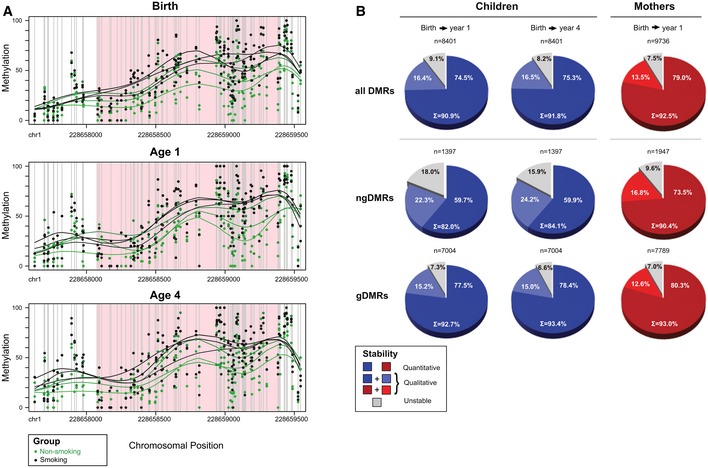
Example of an intergenic ngDMR located 8,965 bp away from the TSS of miR‐466‐6A. Green dots indicate the raw methylation values for non‐smoking samples for all 48 CpGs in the region, while black dots indicate the raw methylation of children from smoking mothers. Lines represent smoothed methylation levels. Methylation differences of 14.5, 13.5, and 12.5%, at time of birth, one year after birth, and four years after birth, respectively, show a strong, quantitative stability of the differential hypermethylation at this locus.
Global analysis over all DMRs, ngDMRs, and gDMRs (from top to bottom row) in mothers and children shows stability of methylation using both quantitative criteria (decrease in absolute mean methylation difference between smokers and non‐smokers should not exceed 5%) as well as qualitative criteria (direction of differential methylation should remain identical irrespective of the absolute methylation change). As expected, genetically determined gDMRs are more stable than their non‐genetically determined counterparts. Still, 90.4% (82–84.1%) of the ngDMRs show longitudinal stability in mothers (children).
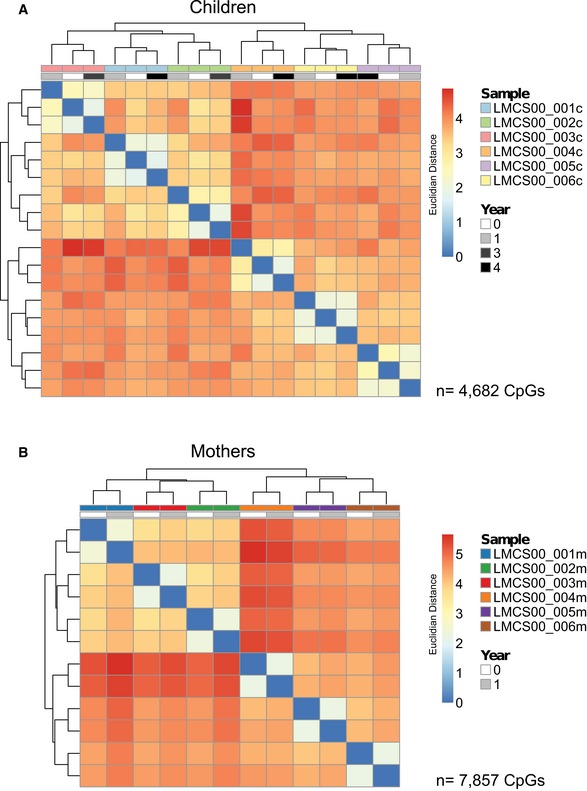
- A, B
Hierarchical clustering (Euclidean distance) of methylation of all CpGs in ngDMRs that are not subject to SNPs in any sample. Only CpGs with a minimum coverage of 10 reads in all samples were considered. Clustering occurs by sample, not by year, suggesting high maintenance of CpG methylation levels for each sample over time. (A) Children (n = 4,682 CpGs), (B) mothers (n = 7,857 CpGs).
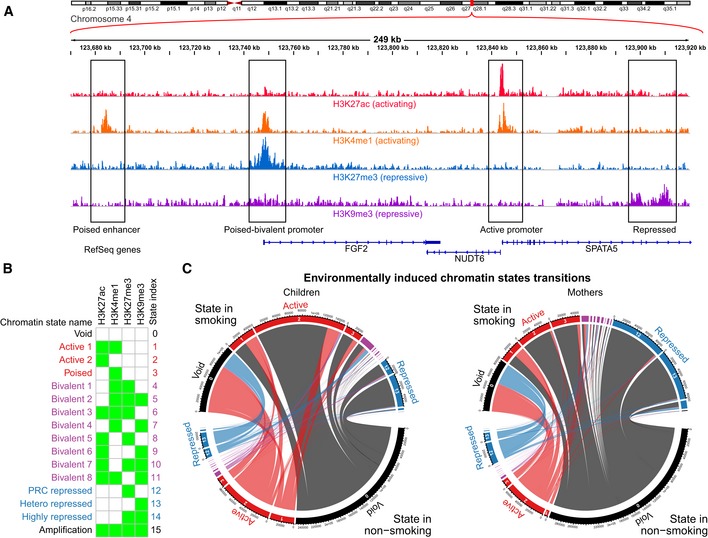
Visualization of four histone mark read densities over 249 kb of chromosome 4 representing active marks (H3K27ac and H3K4me1) and repressive marks (H3K27me3 and H3K9me3). This region illustrates examples of (boxed from left to right): a poised enhancer upstream of FGF2 (exhibiting H3K4me1, but lacking the active signature of H3K27ac); a bivalent promoter of FGF2 (with co‐occurring active H3K4me1 and repressive H3K27ac at the promoter); active promoters marks around the TSS of NUDT6 and SPATA5 (displaying the 2 active marks of H3K27ac and H3K4me1); and repressive marks in the gene body of SPATA5 (H3K9me3), which we observe in the intron of some expressed genes.
Chromatin state model derived from all samples representing all possible 16 states. The states were reorganized and named according to biologically relevant chromatin states representing: lack of signal (state 0, “Void”); active states (states 1–3, in red); bivalent states, showing a combination of both active and repressive marks, organized by the proportion of the genome that they represent on average (states 4–11, in purple); repressed states (states 12–14, in blue); and a state representing co‐occurrence of all marks, most likely representing genomic amplifications (state 15, “Amplification”).
Chord diagram of chromatin state transitions in unstable regions of chromatin. The plot shows the transitions from non‐smoking to smoking for children and mothers, where the size of the outer segments represents the amount of chromatin undergoing transitions from one state to another and the width of the ribbons represents the amount of a single state transiting to another chromatin state. The scale of segment size and ribbon width are comparable between mothers and children. The outer segments and ribbons are colored according to transition to/from void, active, bivalent, repressed, and amplified as black, red, purple, blue, and black, respectively. It can be observed that in children, there is a net overall transition to a more hyperactive chromatin state (i.e., gain of twice as much active chromatin compared to repressed), which is not observed in the mothers who have a more balanced gain of both active and repressive states.
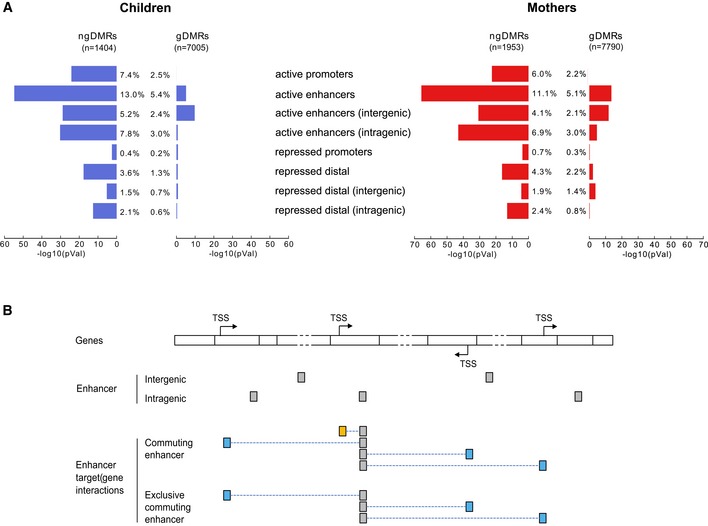
Enrichment of DMRs in annotated chromatin elements for children (left) and mothers (right). The bar plot shows the −log10(P‐value) of the enrichment for ngDMRs (left bars) and gDMRs (right bars). The percentages indicate the proportion of g/ngDMRs that overlap with chromatin elements of the category.
Definitions of various enhancer classes: if at least one gene outside the host gene is regulated, we call it a “commuting” enhancer, as it resides in one gene but act on distal gene(s). When commuting enhancers do not interact with their host gene, we call them “exclusive commuting enhancers”.
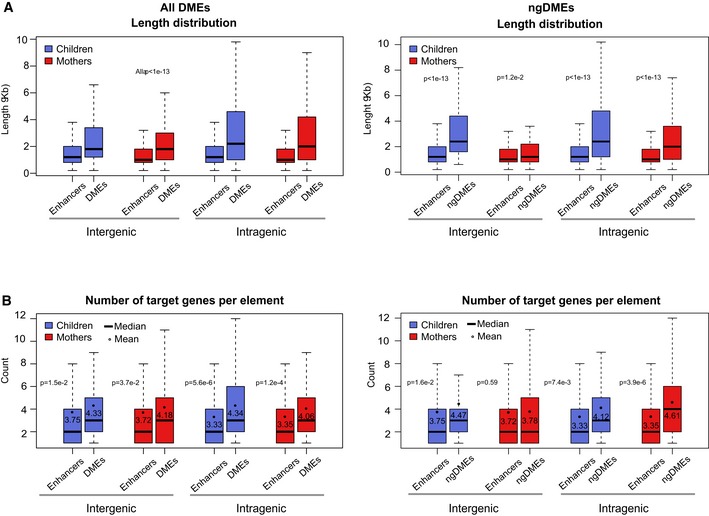
Length distribution of enhancers (non‐TSS‐associated active regulatory regions) and ngDMEs in mothers and children, both inter‐ and intragenic, indicating a significant increase in length for differentially methylated enhancers compared to other enhancers.
Distribution of the number of predicted target genes from the interaction datasets for the same categories as in (A). Black dots indicate the mean number. The corresponding P‐value is indicated (Mann–Whitney test), showing a significant increase for ngDMEs, which is more pronounced in intragenic elements.
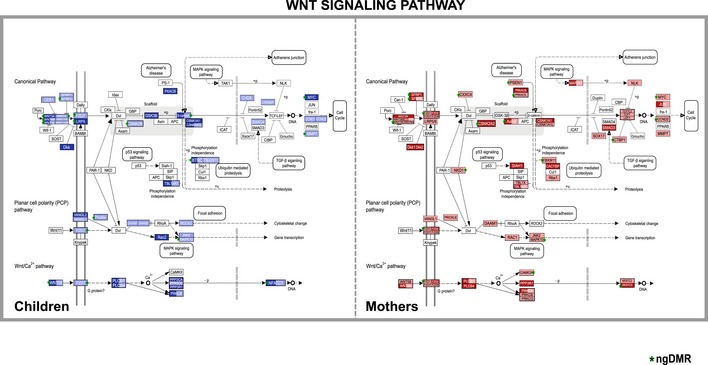
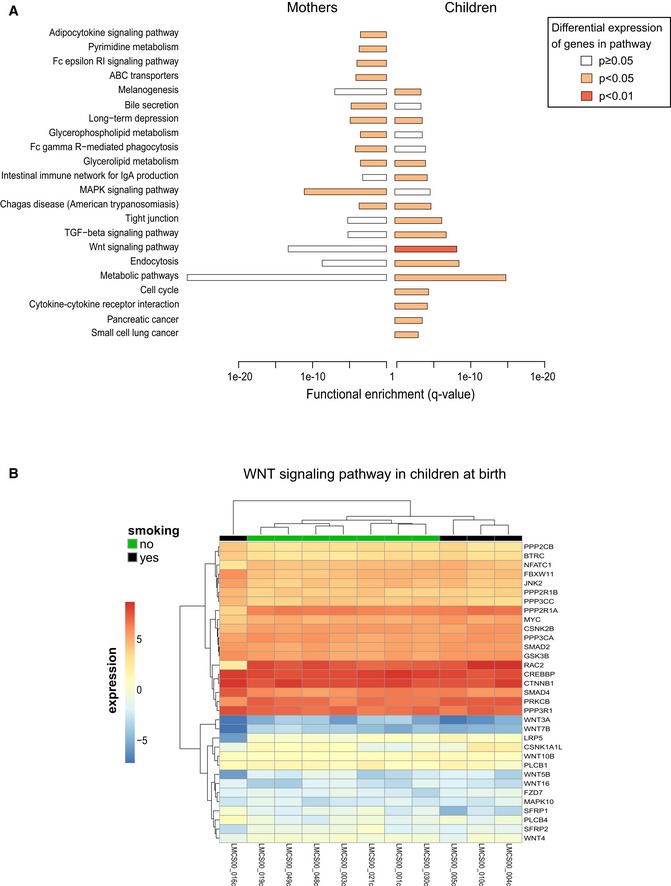
The bar plot shows the list of pathways that are significantly enriched among the target genes of DMRs intersecting active enhancers and promoters both in mothers and in children (enrichment q‐value indicated on x‐axis) and that show differential expression of genes between smoking and non‐smoking samples as measured by RNA‐seq in either children or mothers. Color of bars indicates significance of differential expression of genes in this pathway (see Appendix Supplementary Methods for details of the calculation). Note that the WNT pathway shows the strongest differential expression in children.
WNT signaling genes interacting with differentially methylated enhancers (DMEs) show clustering of smokers versus non‐smokers from the RNA expression profile (DME target prediction was mapped by ChiA‐PET where available and nearest TSS otherwise).
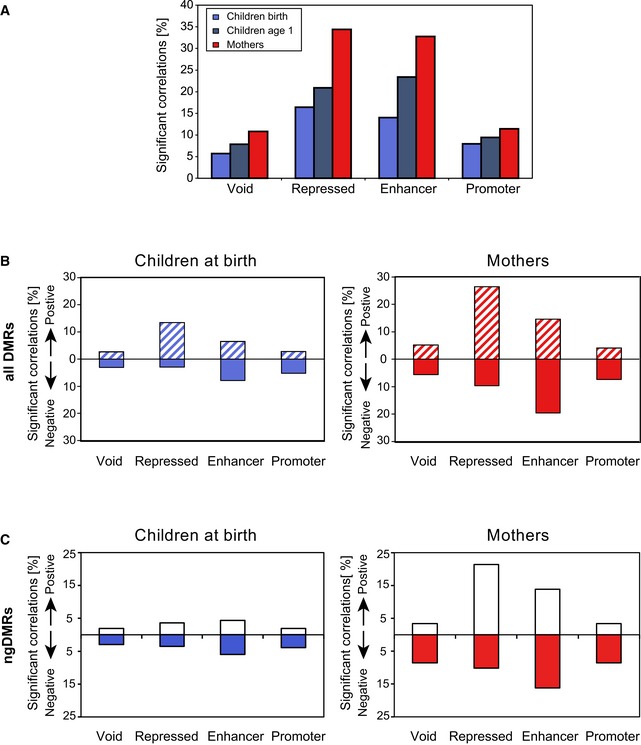
Proportion of DMR‐target gene pairs showing a significant correlation (Spearman's correlation test p < 0.05) between mean methylation and target gene expression, indicating a higher correlation level for DMRs in repressed or enhancer regions as compared to void or promoter regions. Also, correlation levels increase for children from year 0 to year 1 indicating a temporal gap between epigenetic changes and induced gene expression.
Proportion of significantly correlated DMR‐target gene pairs in the same categories as (A), split into positive/negative correlation, showing an excess of positive correlation for DMRs associated with repressed regions (containing one or both PRC‐associated H3K27me3 marks as well as the repressive H3K9me3 marks), and an excess of negative correlations for active regions (enhancers and promoters).
Proportion of significantly correlated ngDMR‐target gene pairs split into positive/negative correlation in the same categories as (B).
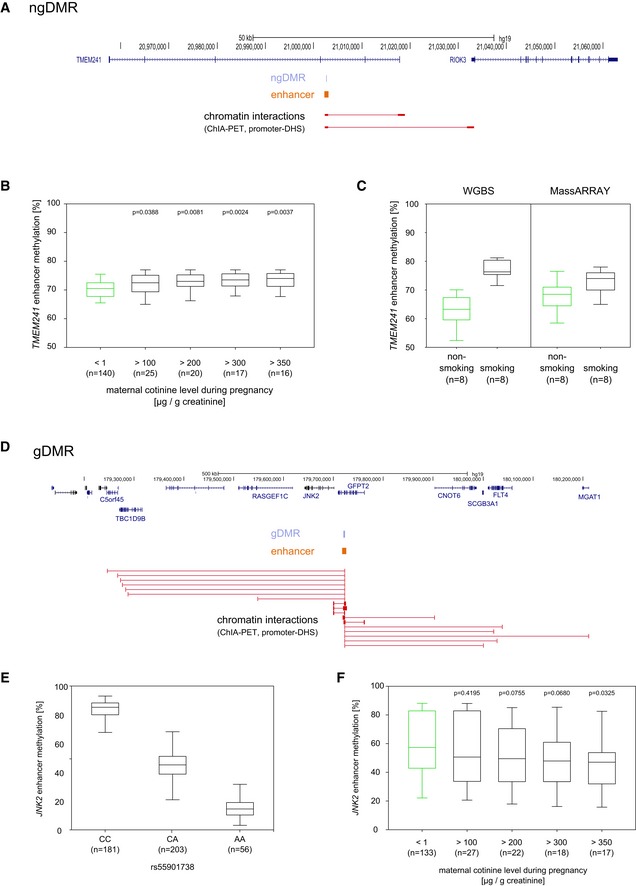
Extended locus of a commuting enhancer overlapping a ngDMR in the TMEM241 intron.
The hypermethylated DMR in the enhancer region identified in the cord blood of prenatally exposed children was confirmed by MassARRAY (n = 471). The results show that concomitant with an increase in maternal cotinine levels, DNA methylation increases in this region.
Box plots compare methylation in the TMEM241 DMR evaluated by WGBS and by MassARRAY, respectively, for the same eight individuals. This comparison suggests that the relatively small effects size observed in the MassARRAY validation seems to be in part related to the weaker spread in methylation values obtained by this method.
Extended locus of the JNK2 enhancer, showing all predicted target genes by ChIA‐PET and promoter‐DHS interaction data.
A clear negative correlation between the genotype of rs55901738 (located inside the JNK2 DME) and the methylation level in this locus was observed in the discovery cohort (N = 181/203/56).
The hypomethylated gDMR within the JNK2 enhancer region was similarly validated by MassARRAY in the entire cohort. Loss of DNA methylation in this region depends on maternal urine cotinine levels and decreases steadily with an increase in maternal urine cotinine concentrations. For children of heavy smoking mothers (cotinine level > 350 ?g/g creatinine) we observe a methylation difference of over 10% compared to non‐smoking mothers.
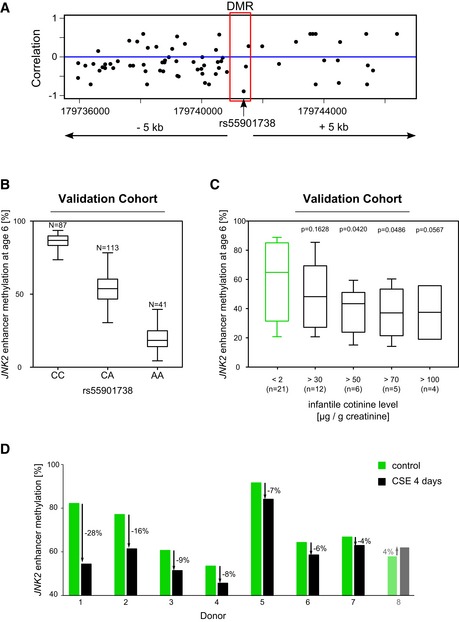
Locus around the JNK2 DMR showing for each genotyped SNP the correlation of the genotype to the methylation in the DMR region.
Genotype at the SNP rs55901738 located inside the DME in the validation cohort (N = 87/113/41) indicating a clear negative correlation between the genotype and the methylation level at this locus.
In an independent validation cohort, we confirmed in six‐year‐old children that cigarette smoke exposure (passive smoking) leads to a decrease of methylation in the JNK2 enhancer region. Children's urine cotinine levels inversely correlate with the JNK2 enhancer methylation level.
Bar plots showing the loss of DNA methylation in the JNK2 enhancer region following a four‐day treatment of PBMCs with cigarette smoke extract in 8 different donors.

Chromatin state comparison between non‐smoking/smoking mothers and their children reveals a differentially active region in the intron of GFPT2, which corresponds to a hypomethylated gDMR in the smoking samples.
Methylation of this region significantly, although weakly, correlates with gene expression of JNK2 (n = 364). Considering children from non‐smoking (urine cotinine [μg]/creatinine [g] < 1, n = 106) or smoking mothers (urine cotinine [μg]/creatinine [g] > 100, n = 21) separately reveals that a significant correlation of methylation and JNK2 transcription is observed only for children of smoking mothers corresponding to the differentially active characteristics of this region.
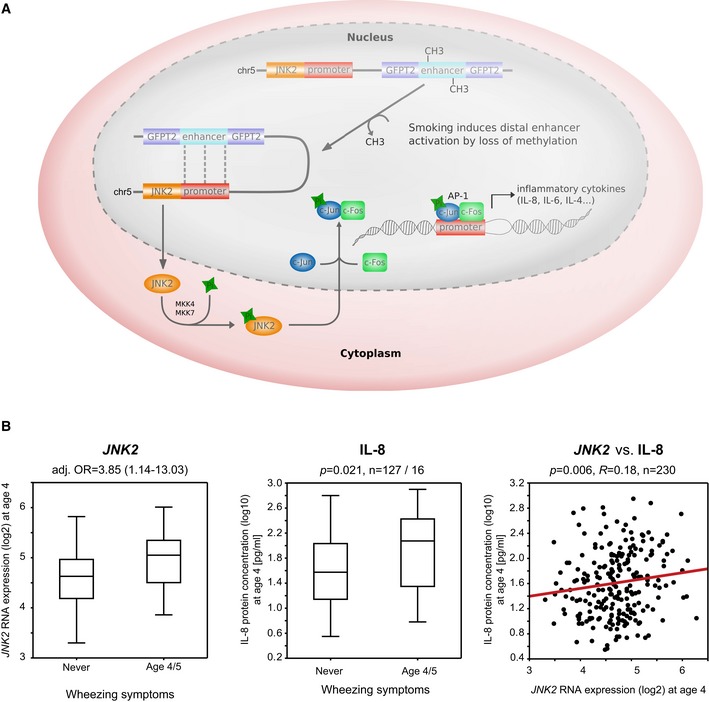
Loss of methylation in the differentially active JNK2 enhancer might contribute to differential JNK2 expression and altered secretion of the cytokine IL‐8. JNK2 as a mediator of the early inflammatory response phosphorylates the transcription factor c‐Jun that subsequently binds c‐Fos, thereby forming the DNA‐binding complex AP‐1 (activator protein 1). AP‐1 binding motifs have been identified in the promoter region of many cyto‐ and chemokines with IL‐8 being one central downstream target.
At age four, JNK2 gene expression and IL‐8 protein concentration (measured in serum) are significantly elevated in wheezing children compared to those without any wheezing or other respiratory symptoms (Mann–Whitney test, P = 0.037 and 0.021, respectively). Both expression patterns correlate significantly in the entire cohort (Spearman's correlation p = 0.006, box plots vizualize 25/75 percentile and median, whiskers represent non‐outlier range).
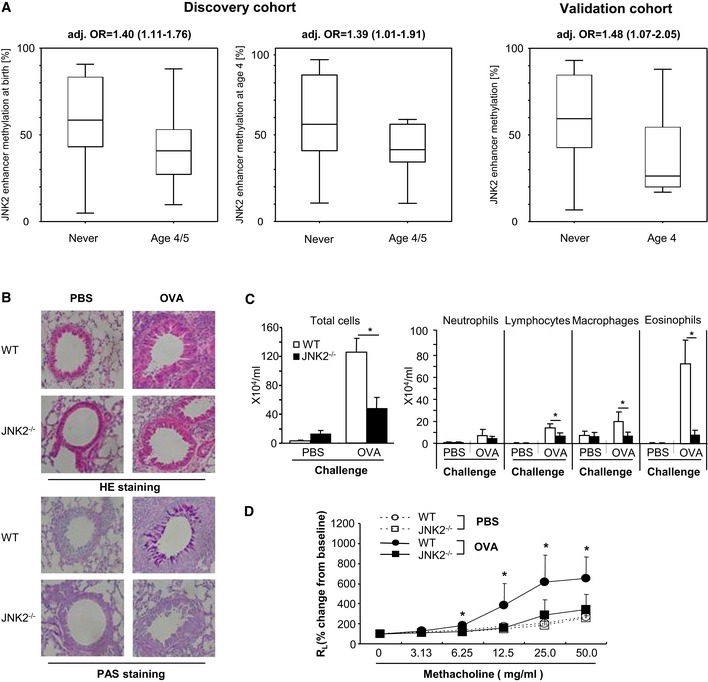
- A
In both the discovery and the validation cohort, we show that loss of DNA methylation in the JNK2 enhancer is associated with an increased risk for the development of wheezing symptoms in four‐year‐old children. Odds ratios are calculated using logistic regression models adjusted for gender, number of siblings, the presence of a cat in the household, parental history of atopy, school education, and smoking during pregnancy (adj. OR). In 4‐year‐old children, regression models were additionally adjusted for postnatal smoking (infantile urine cotinine concentrations, box plots vizualize 25/75 percentile and median, whiskers represent non‐outlier range).
- B–D
JNK2 knockdown leads to a diminished asthma response in OVA‐sensitized mice. (B) HE‐ and PAS‐stained lung sections of OVA‐sensitized JNK2−/− mice show reduced cell infiltration and mucous gland hyperplasia compared to wild‐type controls. (C) The number of eosinophils, lymphocytes, and macrophages in BAL fluid is significantly lower in JNK2−/− mice, (D) which is accompanied by a decrease in AHR compared to OVA‐sensitized WT mice (*P < 0.05 by Student's t‐test, data are shown as mean ± SD (n = 10–15) of three independent experiments).
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
