

Survey of variation in human transcription factors reveals prevalent DNA binding changes
- PMID: 27013732
- PMCID: PMC4825693
- DOI: 10.1126/science.aad2257
Survey of variation in human transcription factors reveals prevalent DNA binding changes
Abstract
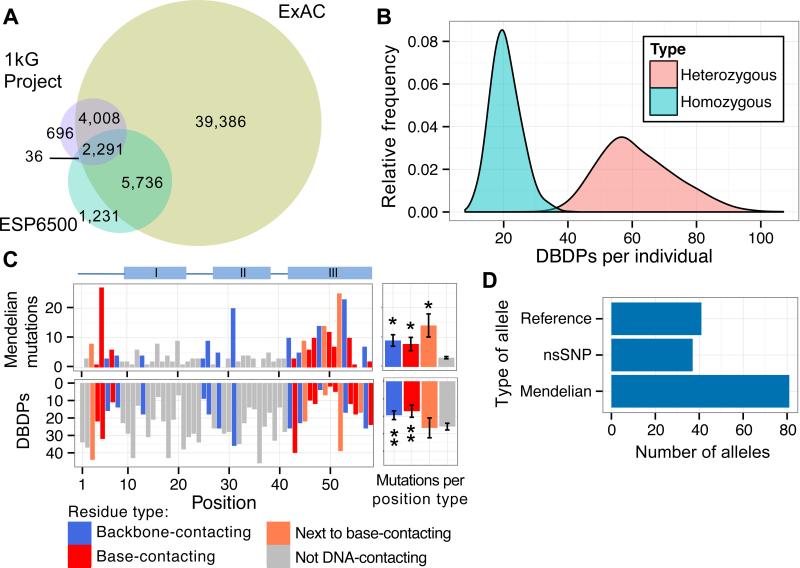
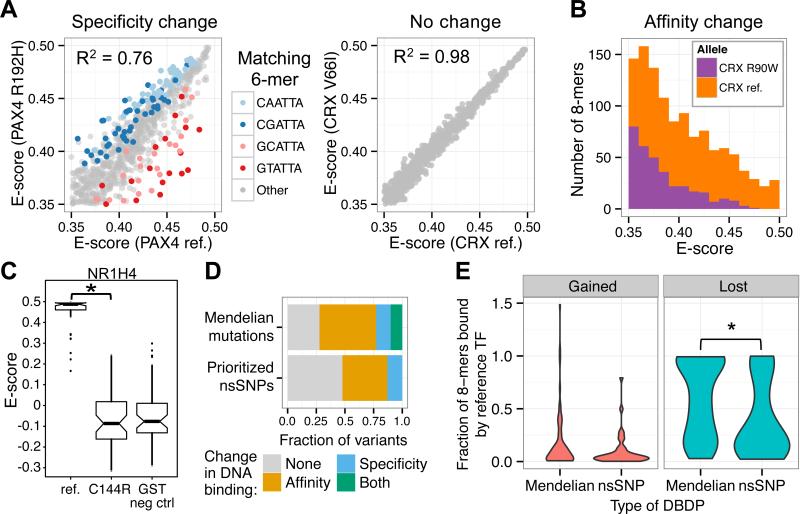
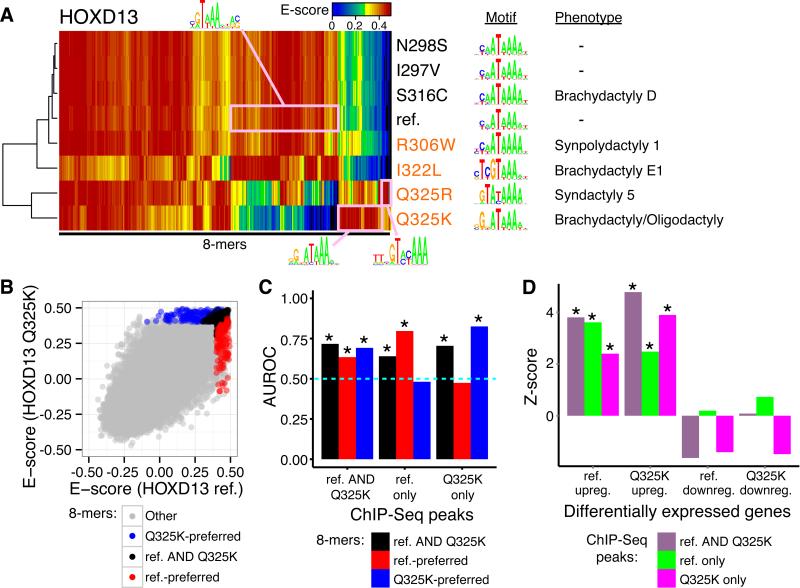
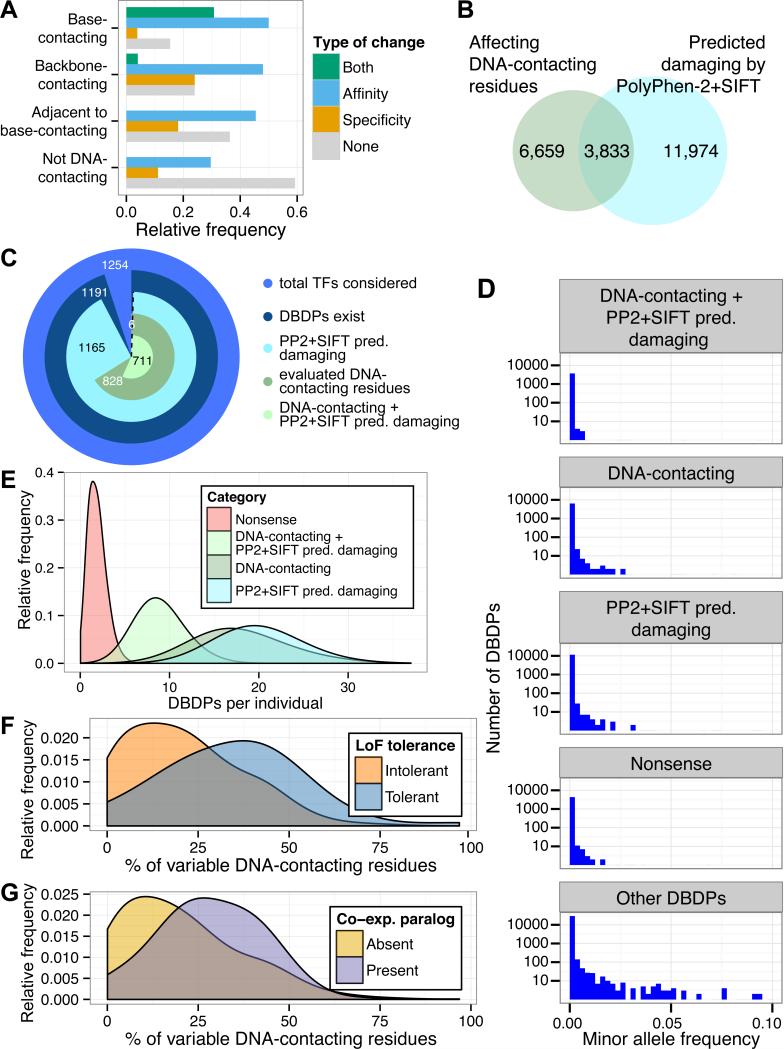
Sequencing of exomes and genomes has revealed abundant genetic variation affecting the coding sequences of human transcription factors (TFs), but the consequences of such variation remain largely unexplored. We developed a computational, structure-based approach to evaluate TF variants for their impact on DNA binding activity and used universal protein-binding microarrays to assay sequence-specific DNA binding activity across 41 reference and 117 variant alleles found in individuals of diverse ancestries and families with Mendelian diseases. We found 77 variants in 28 genes that affect DNA binding affinity or specificity and identified thousands of rare alleles likely to alter the DNA binding activity of human sequence-specific TFs. Our results suggest that most individuals have unique repertoires of TF DNA binding activities, which may contribute to phenotypic variation.
Copyright © 2016, American Association for the Advancement of Science.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
