

Phenylketonuria (PKU): A problem solved?
- PMID: 27014571
- PMCID: PMC4789336
- DOI: 10.1016/j.ymgmr.2015.12.004
Phenylketonuria (PKU): A problem solved?
Abstract
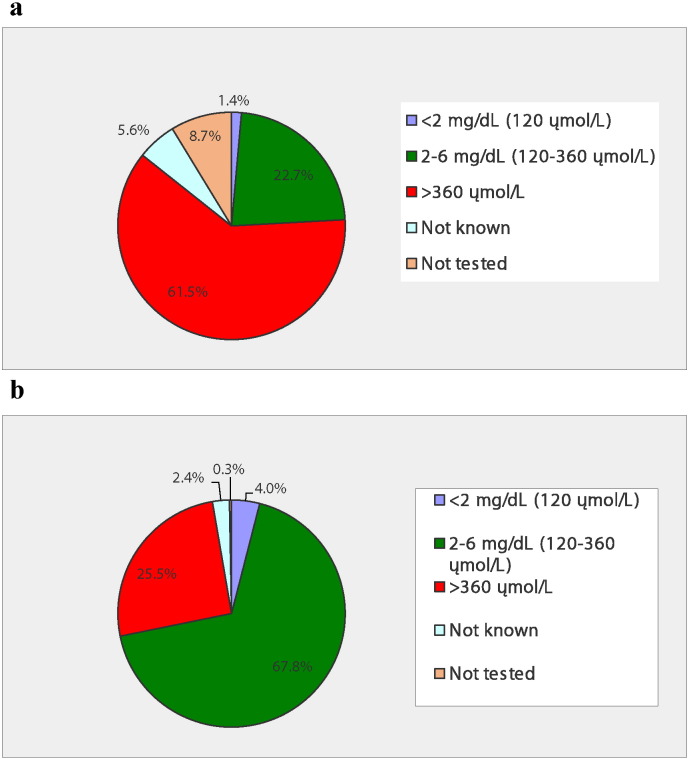
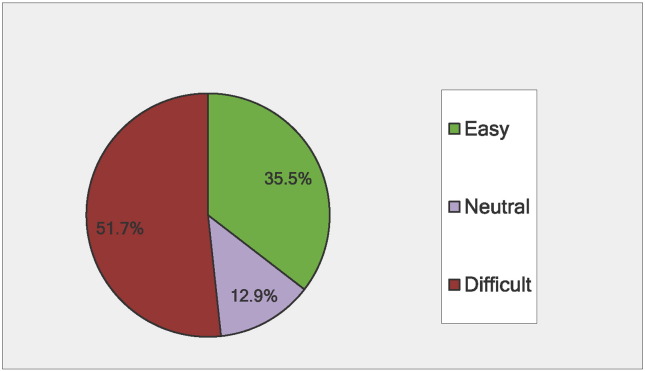
Phenylketonuria (PKU) is a rare metabolic disorder characterized by impaired conversion of phenylalanine (Phe) to tyrosine. If left untreated, the resultant accumulation of excess blood Phe can cause physiological, neurological, and intellectual disabilities. The National PKU Alliance (NPKUA) conducted a survey of its membership to assess current health status and interest in new treatments for PKU. Of the 625 survey respondents, less than half (46.7%) reported blood Phe within (120-360 μmol/L) - the range recommended by the American College of Medical Genetics and Genomics (ACMG). The survey results also showed that younger (≤ 18 years) individuals were about 3-times as successful in keeping their blood Phe concentrations within the recommended clinical range compared with adults. Blood Phe over 360 μmol/L was reported in one-quarter (25.5%) of ≤ 18 year old individuals and almost two-thirds (61.5%) of adults. A little more than half (51.7%) of respondents reported having difficulty in managing their PKU, including the maintenance of a Phe-restricted diet. Individuals with PKU desire new treatments that would allow them to increase their intake of natural protein, discontinue or reduce their intake of medical foods (medical formula and foods modified to be low in protein), improve their mental health (including a reduction in depression and anxiety), and a reduction of their blood Phe concentrations. Respondents preferred oral administration of any newly developed therapies and, in general, disliked therapeutic injections. Injections at home were preferred over injections at a clinic. Payers, government agencies, clinicians, and industry partners should consider patient input when developing and approving new therapies and treatments for PKU.
Keywords: ACMG, American College of Medical Genetics and Genomics; NPKUA, National PKU Alliance; PAH, phenylalanine hydroxylase; PKU, phenylketonuria; Phe, phenylalanine; Phenylalanine; Phenylalanine hydroxylase deficiency; Phenylketonuria.
Figures
References
-
- Phenylketonuria; PKU Online Mendelian Inheritance in Man® omim.org. http://www.omim.org/entry/261600 (last accessed 10-17-15)
-
- Vockley J., Anderson H.C., Antshel K.M., Braverman N.E., Burton B.K., Frazier D.M. For the American College of Medical Genetics and Genomics Therapeutic Committee, phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet. Med. 2014;16:188–200. - PubMed
-
- Wappner R., Cho S., Kronmal R.A., Schuett V., Seashore M.R. Management of phenylketonuria for optimal outcome: a review of guidelines for phenylketonuria management and a report of surveys of parents, patients, and clinic directors. Pediatrics. 1999;104(6):1–9. - PubMed
-
- Longo N., Arnold G.L., Pridjian G., Enns G.M., Ficicioglu C., Parker S. Long-term safety and efficacy of sapropterin: the PKUDOS registry experience. Mol. Genet. Metab. 2015;114:557–563. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
